

Lysine demethylase KDM2A inhibits TET2 to promote DNA methylation and silencing of tumor suppressor genes in breast cancer
- PMID: 28785073
- PMCID: PMC5608919
- DOI: 10.1038/oncsis.2017.71
Lysine demethylase KDM2A inhibits TET2 to promote DNA methylation and silencing of tumor suppressor genes in breast cancer
Abstract
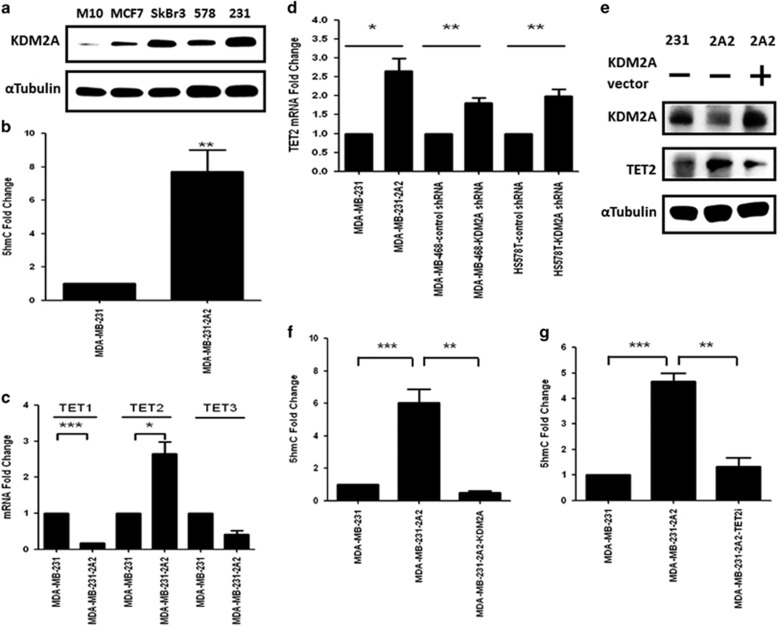
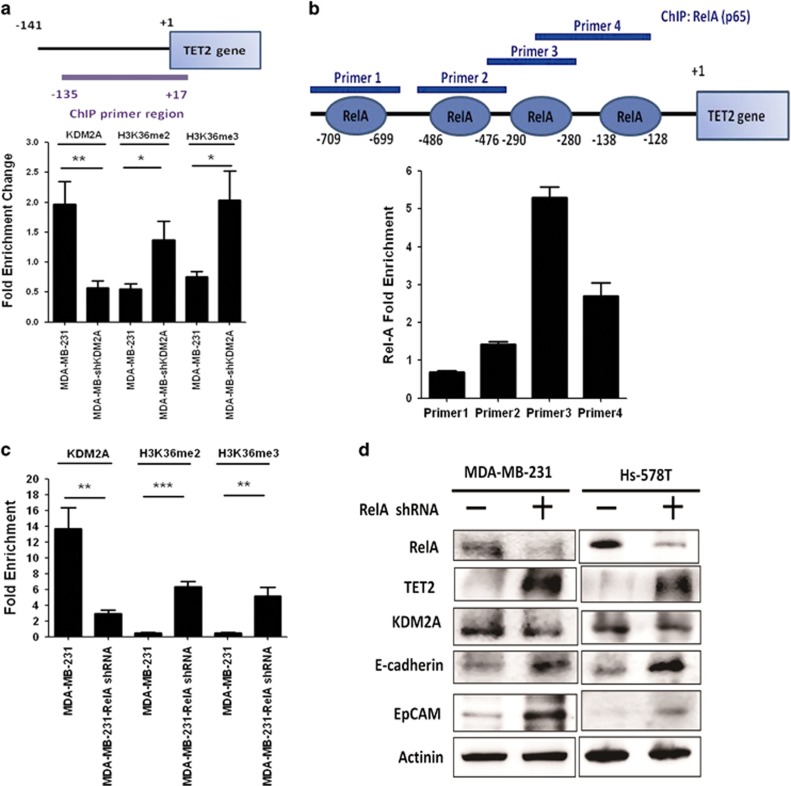
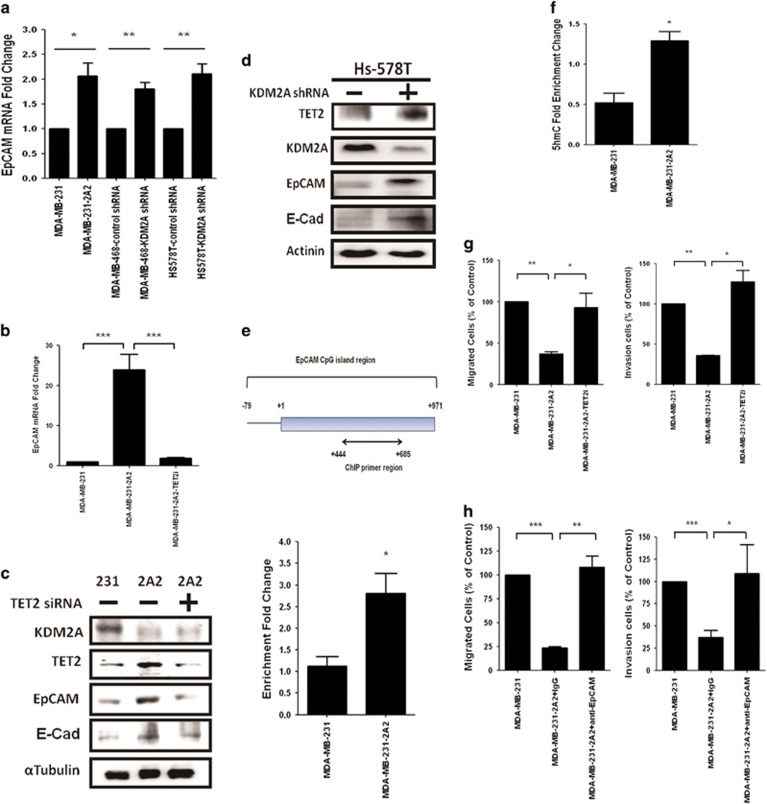
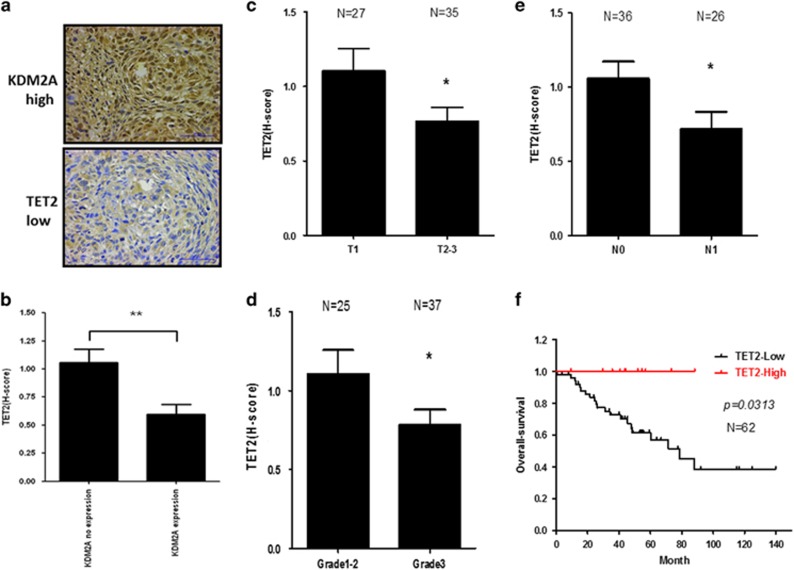
The coupling between DNA methylation and histone modification contributes to aberrant expression of oncogenes or tumor suppressor genes that leads to tumor development. Our previous study demonstrated that lysine demethylase 2A (KDM2A) functions as an oncogene in breast cancer by promoting cancer stemness and angiogenesis via activation of the Notch signaling. Here, we demonstrate that knockdown of KDM2A significantly increases the 5'-hydroxymethylcytosine (5'-hmc) level in genomic DNA and expression of tet-eleven translocation 2 (TET2) in various breast cancer cell lines. Conversely, ectopic expression of KDM2A inhibits TET2 expression in KDM2A-depleted cells suggesting TET2 is a transcriptional repression target of KDM2A. Our results show that KDM2A interacts with RelA to co-occupy at the TET2 gene promoter to repress transcription and depletion of RelA or KDM2A restores TET2 expression. Upregulation of TET2 in the KDM2A-depleted cells induces the re-activation of two TET downstream tumor suppressor genes, epithelial cell adhesion molecule (EpCAM) and E-cadherin, and inhibits migration and invasion. On the contrary, knockdown of TET2 in these cells decreases EpCAM and E-cadherin and increases cell invasiveness. More importantly, TET2 expression is negatively associated KDM2A in triple-negative breast tumor tissues, and its expression predicts a better survival. Taken together, we demonstrate for the first time that TET2 is a direct repression target of KDM2A and reveal a novel mechanism by which KDM2A promotes DNA methylation and breast cancer progression via the inhibition of a DNA demethylase.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10: 295–304. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
