

Statistical Analysis on the Performance of Molecular Mechanics Poisson-Boltzmann Surface Area versus Absolute Binding Free Energy Calculations: Bromodomains as a Case Study
- PMID: 28786670
- PMCID: PMC5615372
- DOI: 10.1021/acs.jcim.7b00347
Statistical Analysis on the Performance of Molecular Mechanics Poisson-Boltzmann Surface Area versus Absolute Binding Free Energy Calculations: Bromodomains as a Case Study
Abstract
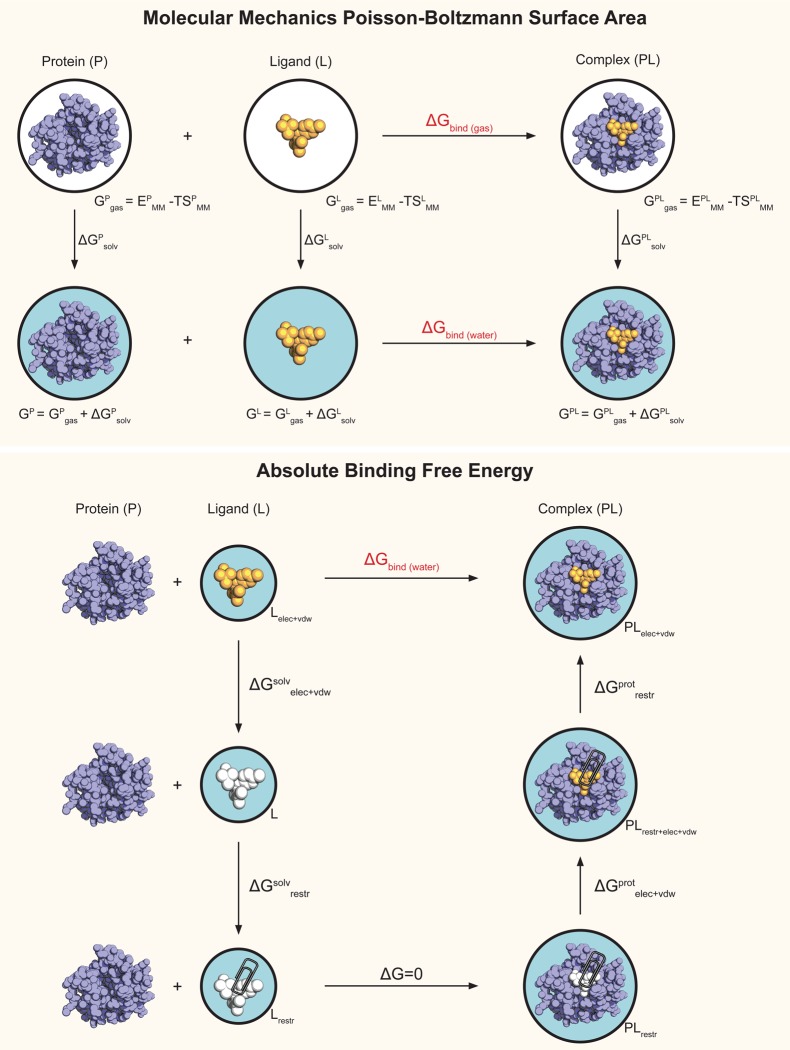
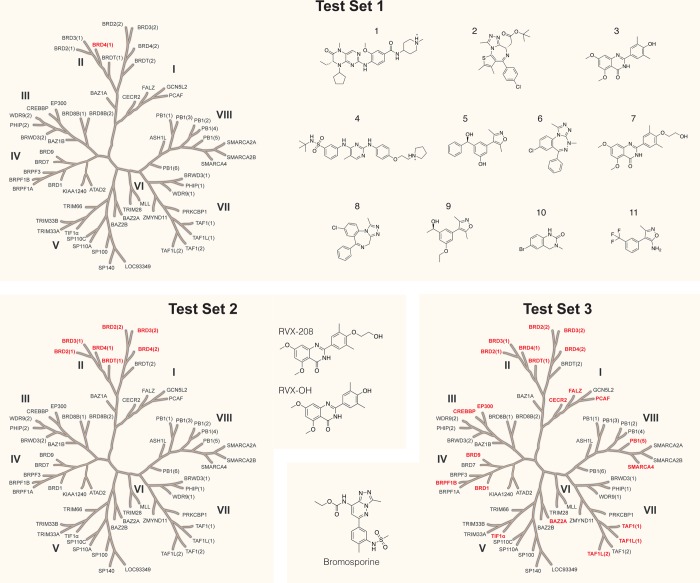
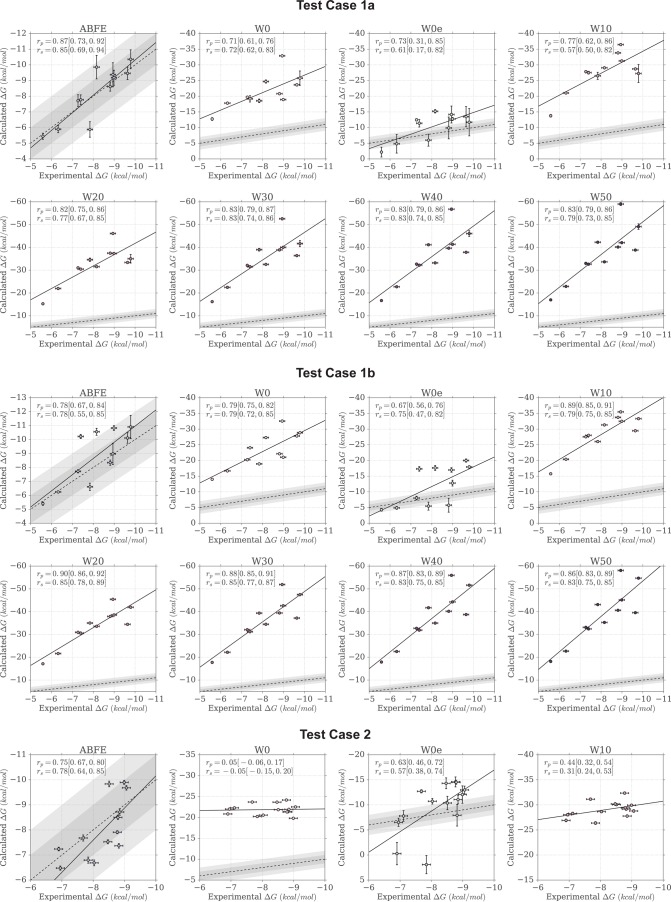
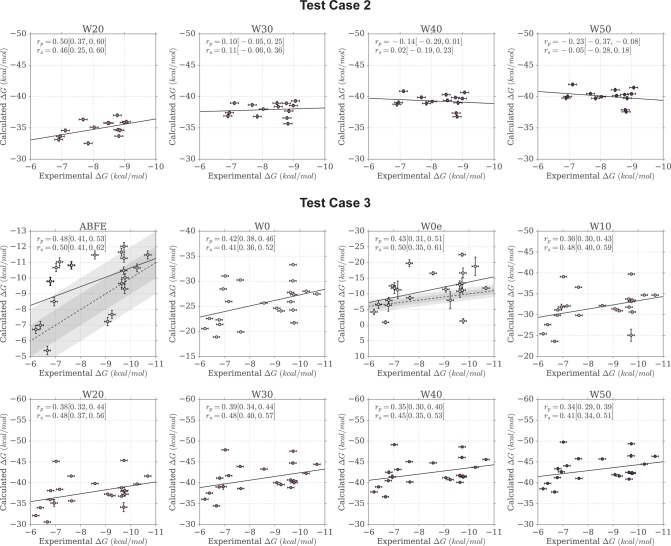
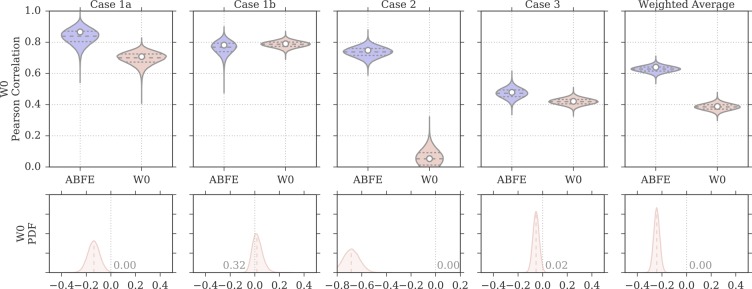
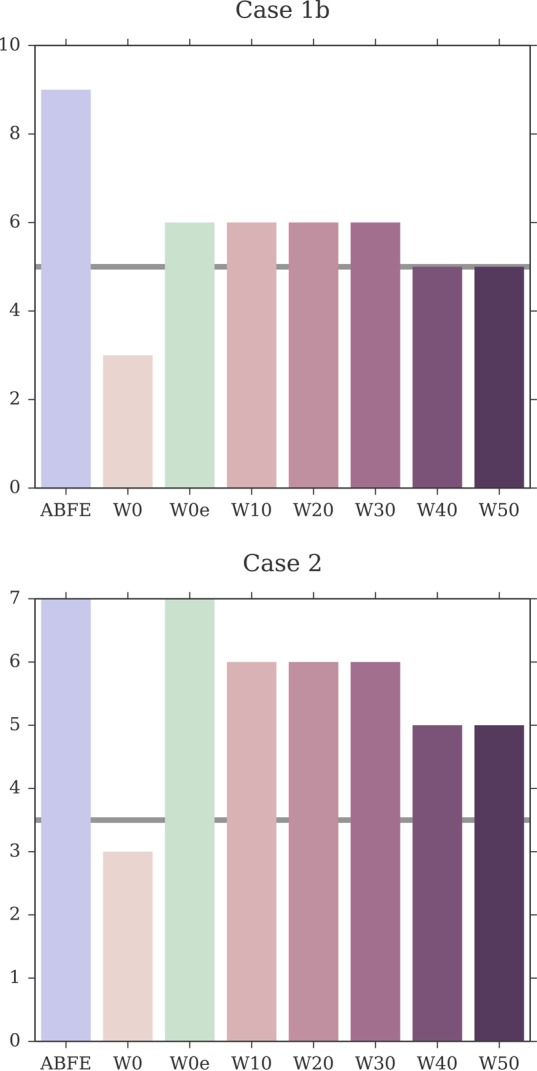
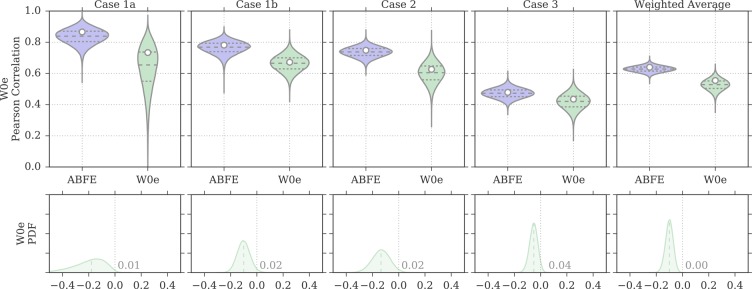
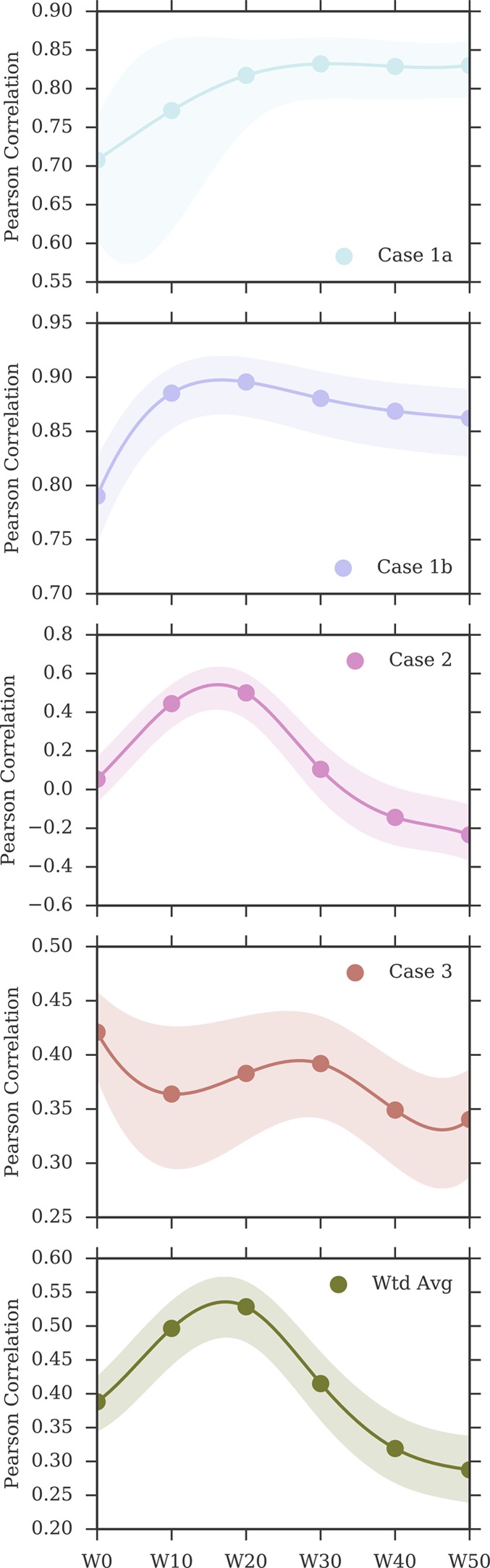
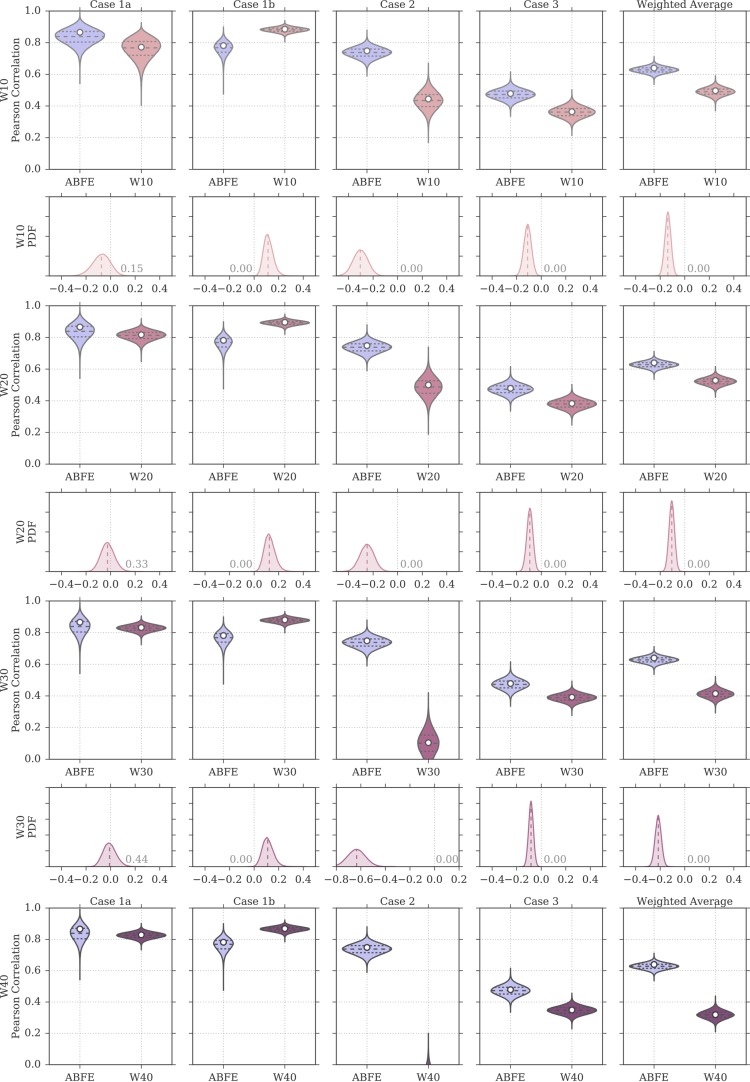
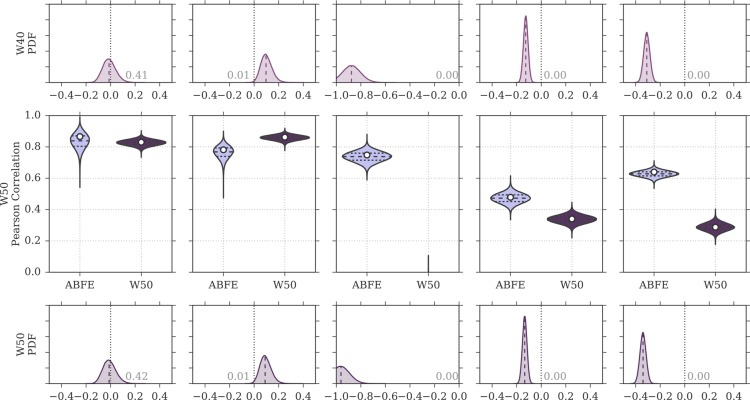
Binding free energy calculations that make use of alchemical pathways are becoming increasingly feasible thanks to advances in hardware and algorithms. Although relative binding free energy (RBFE) calculations are starting to find widespread use, absolute binding free energy (ABFE) calculations are still being explored mainly in academic settings due to the high computational requirements and still uncertain predictive value. However, in some drug design scenarios, RBFE calculations are not applicable and ABFE calculations could provide an alternative. Computationally cheaper end-point calculations in implicit solvent, such as molecular mechanics Poisson-Boltzmann surface area (MMPBSA) calculations, could too be used if one is primarily interested in a relative ranking of affinities. Here, we compare MMPBSA calculations to previously performed absolute alchemical free energy calculations in their ability to correlate with experimental binding free energies for three sets of bromodomain-inhibitor pairs. Different MMPBSA approaches have been considered, including a standard single-trajectory protocol, a protocol that includes a binding entropy estimate, and protocols that take into account the ligand hydration shell. Despite the improvements observed with the latter two MMPBSA approaches, ABFE calculations were found to be overall superior in obtaining correlation with experimental affinities for the test cases considered. A difference in weighted average Pearson ([Formula: see text]) and Spearman ([Formula: see text]) correlations of 0.25 and 0.31 was observed when using a standard single-trajectory MMPBSA setup ([Formula: see text] = 0.64 and [Formula: see text] = 0.66 for ABFE; [Formula: see text] = 0.39 and [Formula: see text] = 0.35 for MMPBSA). The best performing MMPBSA protocols returned weighted average Pearson and Spearman correlations that were about 0.1 inferior to ABFE calculations: [Formula: see text] = 0.55 and [Formula: see text] = 0.56 when including an entropy estimate, and [Formula: see text] = 0.53 and [Formula: see text] = 0.55 when including explicit water molecules. Overall, the study suggests that ABFE calculations are indeed the more accurate approach, yet there is also value in MMPBSA calculations considering the lower compute requirements, and if agreement to experimental affinities in absolute terms is not of interest. Moreover, for the specific protein-ligand systems considered in this study, we find that including an explicit ligand hydration shell or a binding entropy estimate in the MMPBSA calculations resulted in significant performance improvements at a negligible computational cost.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Larsson P.; Hess B.; Lindahl E. Algorithm Improvements for Molecular Dynamics Simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 93–108. 10.1002/wcms.3. - DOI
-
- Shirts M. R.; Mobley D. L.; Brown S. P.. Free-Energy Calculations in Structure-Based Drug Design. In Drug Design; Merz K. M.; Ringe D.; Reynolds C. H., Eds.; Cambridge University Press, 2010; pp 61–86.
-
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. 10.1021/ar000033j. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
