

Computational-experimental approach to drug-target interaction mapping: A case study on kinase inhibitors
- PMID: 28787438
- PMCID: PMC5560747
- DOI: 10.1371/journal.pcbi.1005678
Computational-experimental approach to drug-target interaction mapping: A case study on kinase inhibitors
Abstract
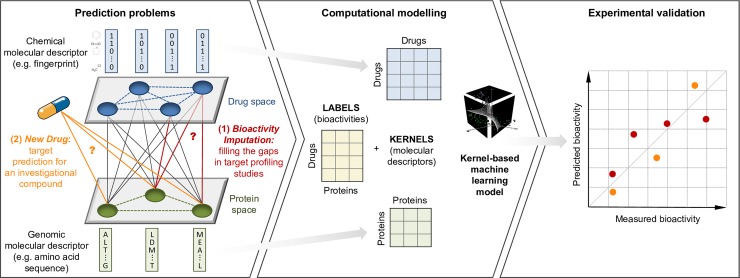
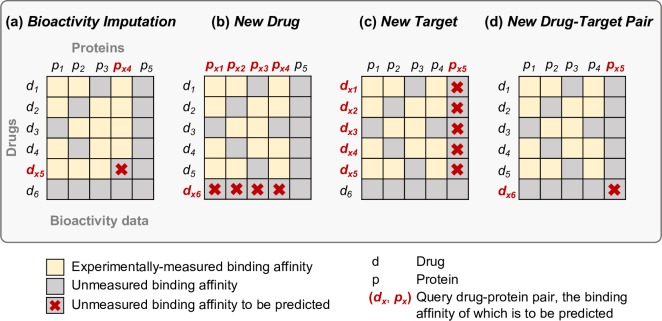
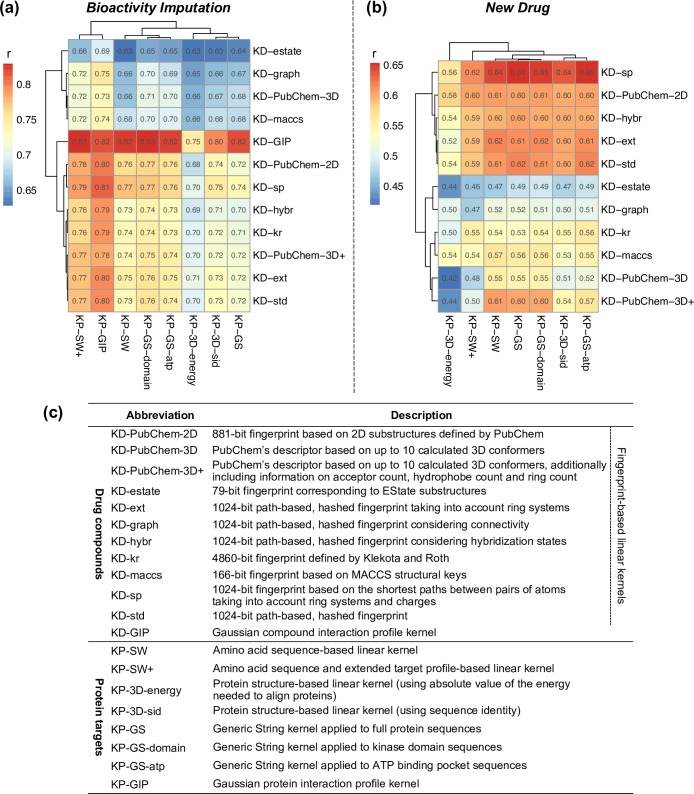
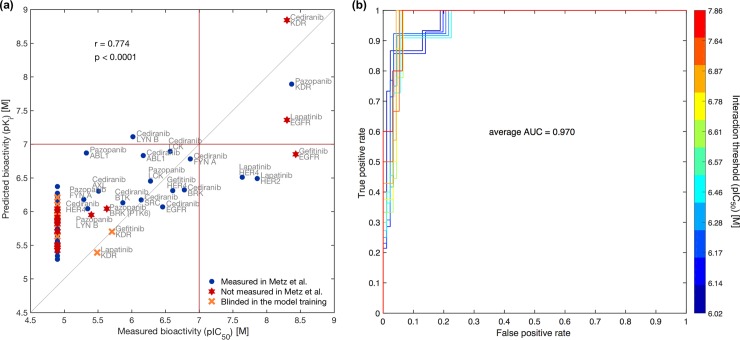
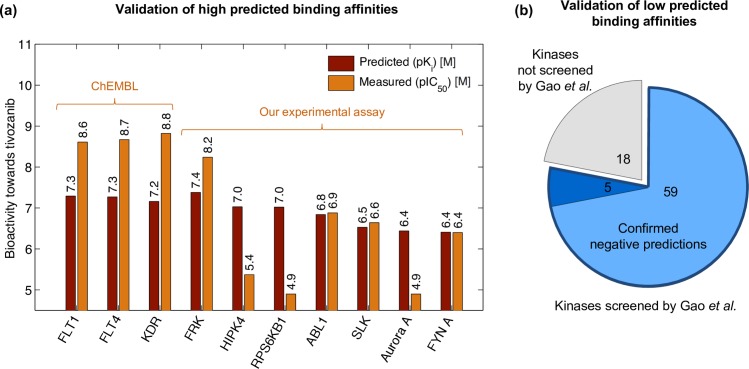
Due to relatively high costs and labor required for experimental profiling of the full target space of chemical compounds, various machine learning models have been proposed as cost-effective means to advance this process in terms of predicting the most potent compound-target interactions for subsequent verification. However, most of the model predictions lack direct experimental validation in the laboratory, making their practical benefits for drug discovery or repurposing applications largely unknown. Here, we therefore introduce and carefully test a systematic computational-experimental framework for the prediction and pre-clinical verification of drug-target interactions using a well-established kernel-based regression algorithm as the prediction model. To evaluate its performance, we first predicted unmeasured binding affinities in a large-scale kinase inhibitor profiling study, and then experimentally tested 100 compound-kinase pairs. The relatively high correlation of 0.77 (p < 0.0001) between the predicted and measured bioactivities supports the potential of the model for filling the experimental gaps in existing compound-target interaction maps. Further, we subjected the model to a more challenging task of predicting target interactions for such a new candidate drug compound that lacks prior binding profile information. As a specific case study, we used tivozanib, an investigational VEGF receptor inhibitor with currently unknown off-target profile. Among 7 kinases with high predicted affinity, we experimentally validated 4 new off-targets of tivozanib, namely the Src-family kinases FRK and FYN A, the non-receptor tyrosine kinase ABL1, and the serine/threonine kinase SLK. Our sub-sequent experimental validation protocol effectively avoids any possible information leakage between the training and validation data, and therefore enables rigorous model validation for practical applications. These results demonstrate that the kernel-based modeling approach offers practical benefits for probing novel insights into the mode of action of investigational compounds, and for the identification of new target selectivities for drug repurposing applications.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010; 10:130–7. doi: 10.1038/nrc2787 - DOI - PMC - PubMed
-
- Hu Y, Furtmann N, Bajorath J. Current compound coverage of the kinome: miniperspective. J Med Chem. 2014; 58:30–40. doi: 10.1021/jm5008159 - DOI - PubMed
-
- Metz JT, Johnson EF, Soni NB, Merta PJ, Kifle L, Hajduk PJ. Navigating the kinome. Nat Chem Biol. 2011; 7:200–2. doi: 10.1038/nchembio.530 - DOI - PubMed
-
- Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014; 346:1255784 doi: 10.1126/science.1255784 - DOI - PubMed
-
- Reymond JL, Awale M. Exploring chemical space for drug discovery using the chemical universe database. ACS Chem Neurosci. 2012; 3:649–57. doi: 10.1021/cn3000422 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
