

Splicing and transcription touch base: co-transcriptional spliceosome assembly and function
- PMID: 28792005
- PMCID: PMC5928008
- DOI: 10.1038/nrm.2017.63
Splicing and transcription touch base: co-transcriptional spliceosome assembly and function
Abstract
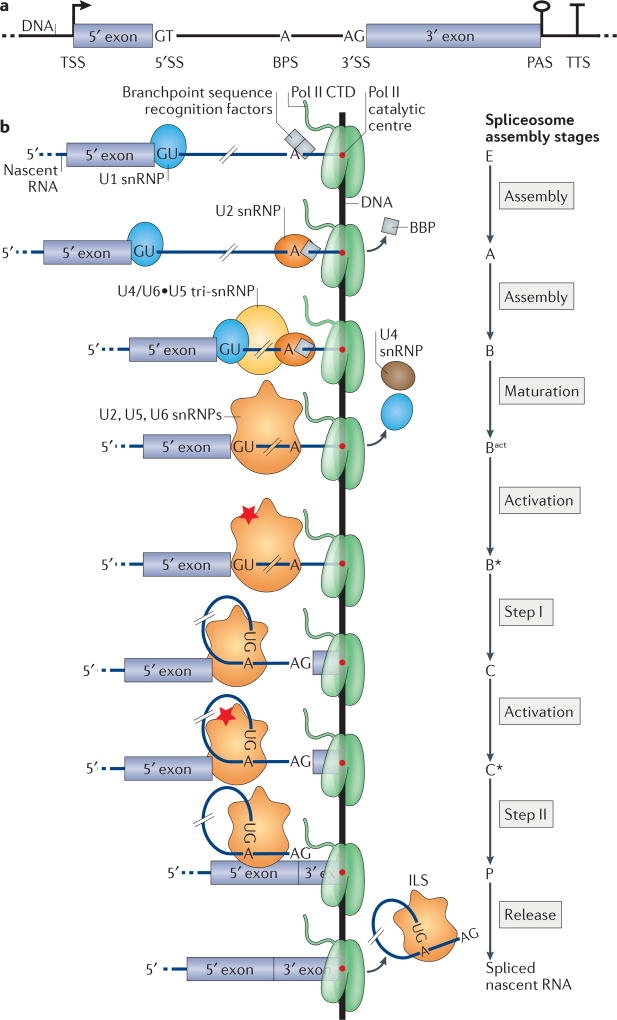
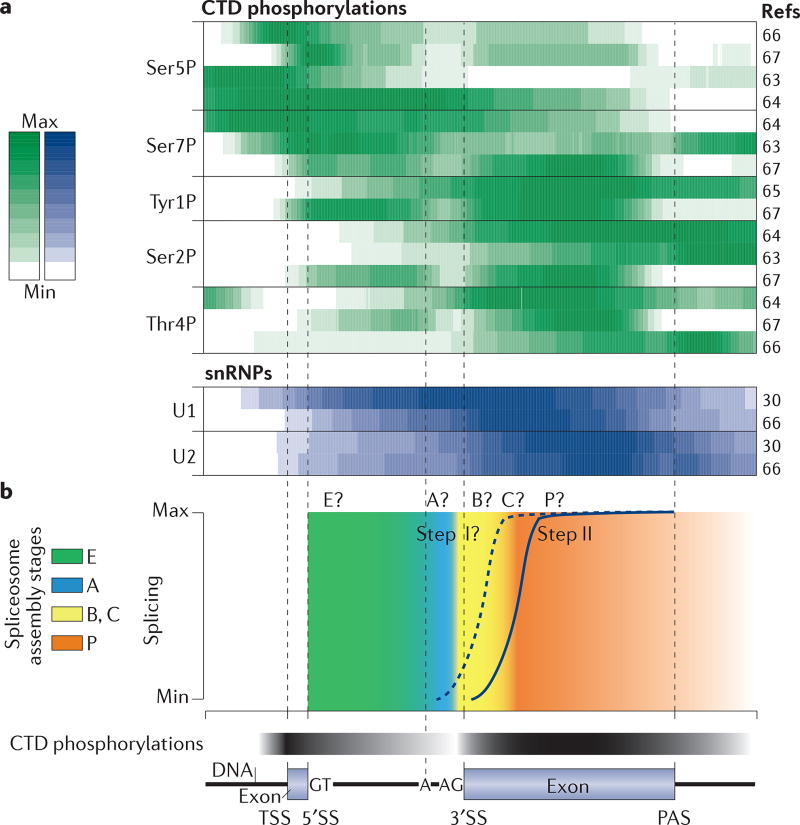
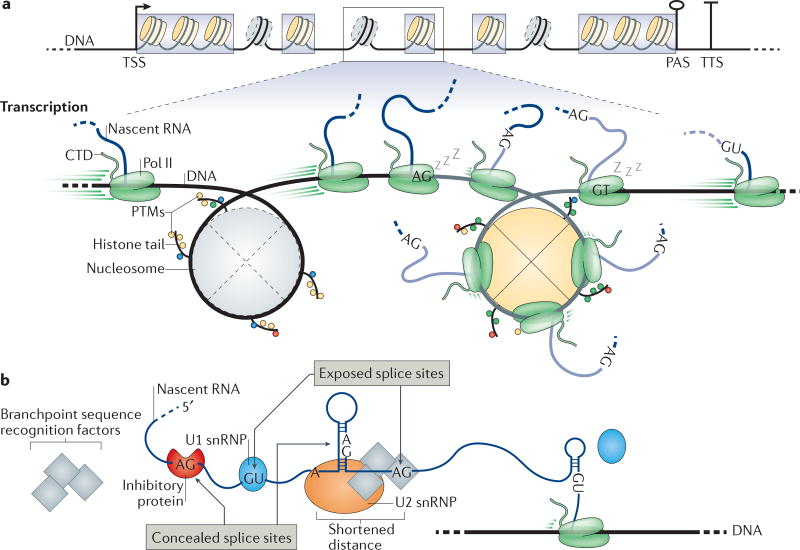
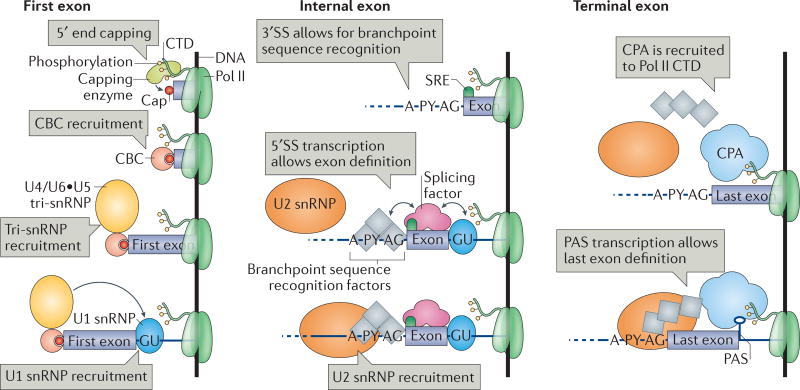
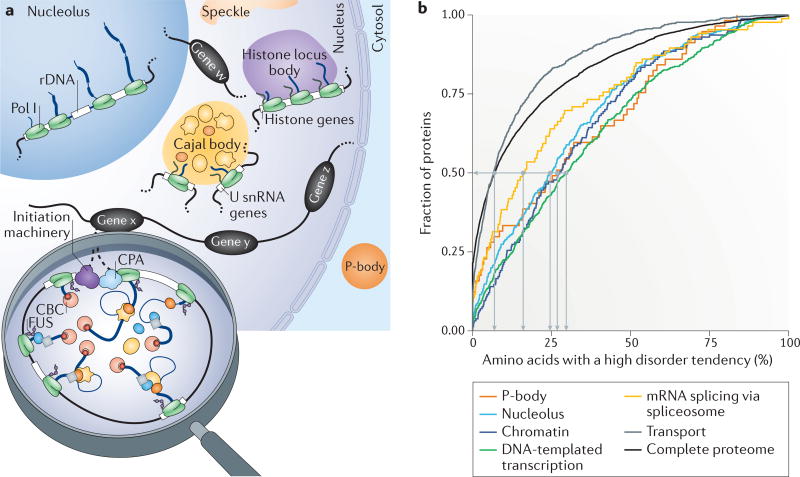
Several macromolecular machines collaborate to produce eukaryotic messenger RNA. RNA polymerase II (Pol II) translocates along genes that are up to millions of base pairs in length and generates a flexible RNA copy of the DNA template. This nascent RNA harbours introns that are removed by the spliceosome, which is a megadalton ribonucleoprotein complex that positions the distant ends of the intron into its catalytic centre. Emerging evidence that the catalytic spliceosome is physically close to Pol II in vivo implies that transcription and splicing occur on similar timescales and that the transcription and splicing machineries may be spatially constrained. In this Review, we discuss aspects of spliceosome assembly, transcription elongation and other co-transcriptional events that allow the temporal coordination of co-transcriptional splicing.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
