

Equine skeletal muscle adaptations to exercise and training: evidence of differential regulation of autophagosomal and mitochondrial components
- PMID: 28793853
- PMCID: PMC5551008
- DOI: 10.1186/s12864-017-4007-9
Equine skeletal muscle adaptations to exercise and training: evidence of differential regulation of autophagosomal and mitochondrial components
Abstract
Background: A single bout of exercise induces changes in gene expression in skeletal muscle. Regular exercise results in an adaptive response involving changes in muscle architecture and biochemistry, and is an effective way to manage and prevent common human diseases such as obesity, cardiovascular disorders and type II diabetes. However, the biomolecular mechanisms underlying such responses still need to be fully elucidated. Here we performed a transcriptome-wide analysis of skeletal muscle tissue in a large cohort of untrained Thoroughbred horses (n = 51) before and after a bout of high-intensity exercise and again after an extended period of training. We hypothesized that regular high-intensity exercise training primes the transcriptome for the demands of high-intensity exercise.
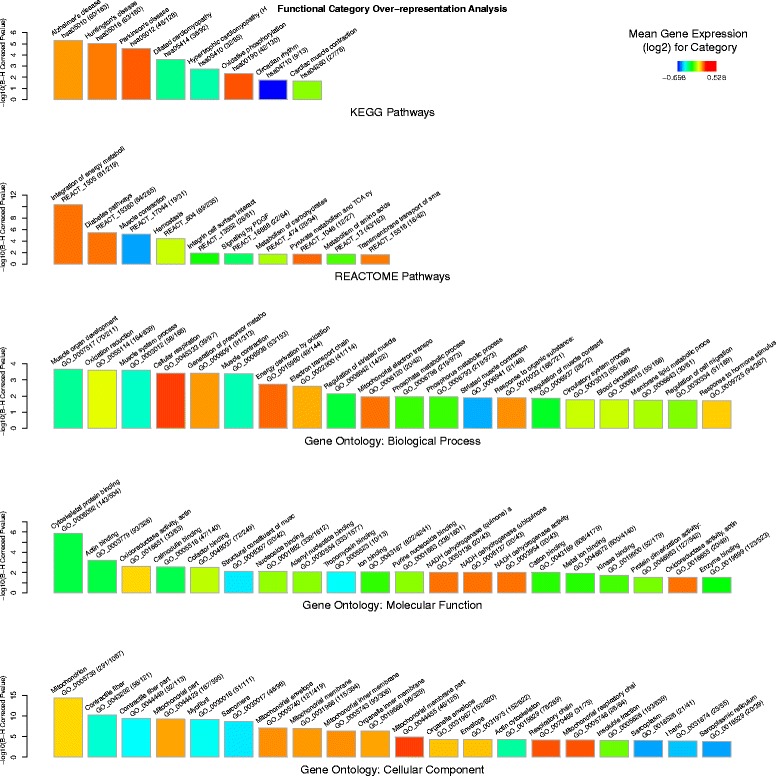
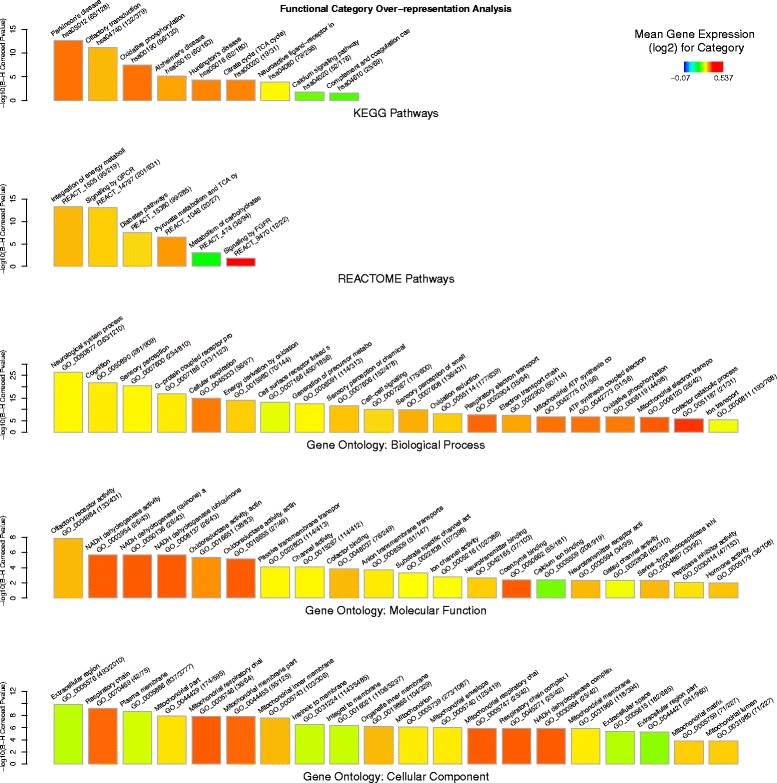
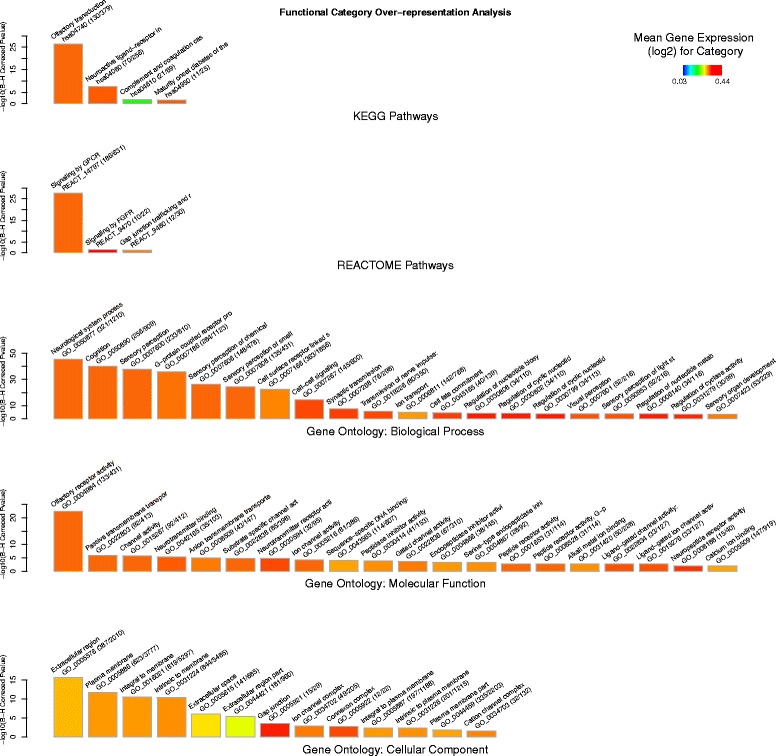
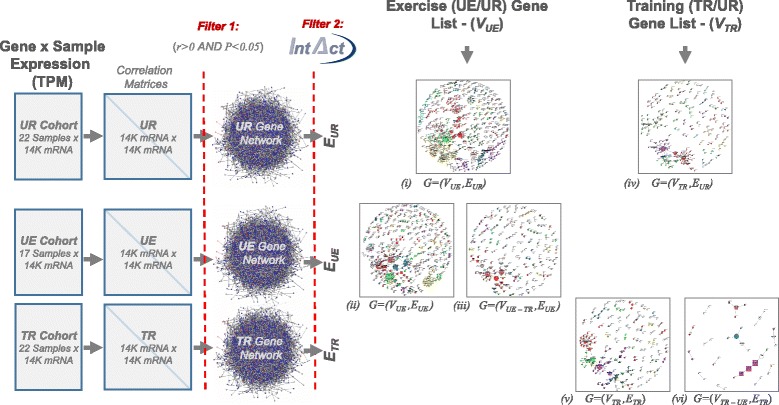
Results: An extensive set of genes was observed to be significantly differentially regulated in response to a single bout of high-intensity exercise in the untrained cohort (3241 genes) and following multiple bouts of high-intensity exercise training over a six-month period (3405 genes). Approximately one-third of these genes (1025) and several biological processes related to energy metabolism were common to both the exercise and training responses. We then developed a novel network-based computational analysis pipeline to test the hypothesis that these transcriptional changes also influence the contextual molecular interactome and its dynamics in response to exercise and training. The contextual network analysis identified several important hub genes, including the autophagosomal-related gene GABARAPL1, and dynamic functional modules, including those enriched for mitochondrial respiratory chain complexes I and V, that were differentially regulated and had their putative interactions 're-wired' in the exercise and/or training responses.
Conclusion: Here we have generated for the first time, a comprehensive set of genes that are differentially expressed in Thoroughbred skeletal muscle in response to both exercise and training. These data indicate that consecutive bouts of high-intensity exercise result in a priming of the skeletal muscle transcriptome for the demands of the next exercise bout. Furthermore, this may also lead to an extensive 're-wiring' of the molecular interactome in both exercise and training and include key genes and functional modules related to autophagy and the mitochondrion.
Keywords: Autophagy; Equine; Exercise; Functional module; Mitochondria; Network analysis; RNAseq; Skeletal muscle; Training; Transcriptome.
Conflict of interest statement
Consent for publication
All authors read and approved the final manuscript
Competing interests
None of the authors has any financial or personal relationships that could inappropriately influence or bias the content of the paper. EWH and DEM are shareholders in Plusvital Ltd., an equine nutrition and genetic testing company. Plusvital Ltd. has been granted a license for commercial use of data contained within patent applications: United States Provisional Serial Number 61/136553 and Irish patent application number 2008/0735, Patent Cooperation Treaty filing: A method for predicting athletic performance potential, September 7, 2009. EWH, DEM and LMK are named on the applications. The patent contents are not related to this manuscript. Plusvital Ltd. had no part in the research in the manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures



References
-
- Hinchcliff KW, Kaneps AJ, Geor RJ. Equine exercise physiology: the science of exercise in the athletic horse. New York. Edinburgh: Saunders/Elsevier; 2008.
-
- Dingboom EGDG, Enzerink E, van Oudheusden HC, Weijs WA. Postnatal muscle fibre composition of the gluteus medius muscle of Dutch Warmblood foals; maturation and the influence of exercise. Equine Vet J. 1999;31(Suppl):95–100. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
