

Annotating pathogenic non-coding variants in genic regions
- PMID: 28794409
- PMCID: PMC5550444
- DOI: 10.1038/s41467-017-00141-2
Annotating pathogenic non-coding variants in genic regions
Abstract
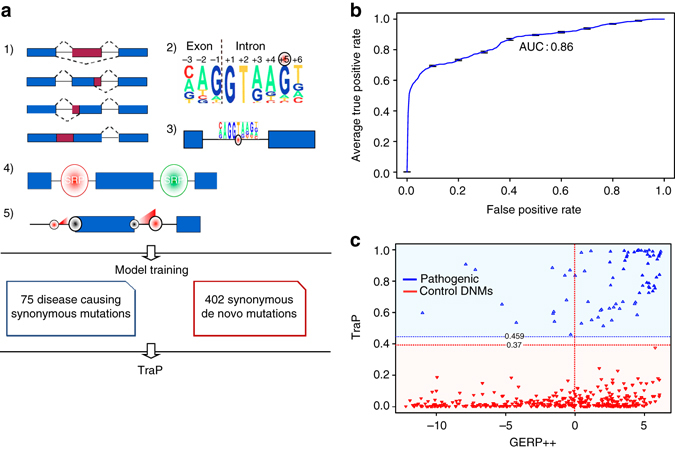
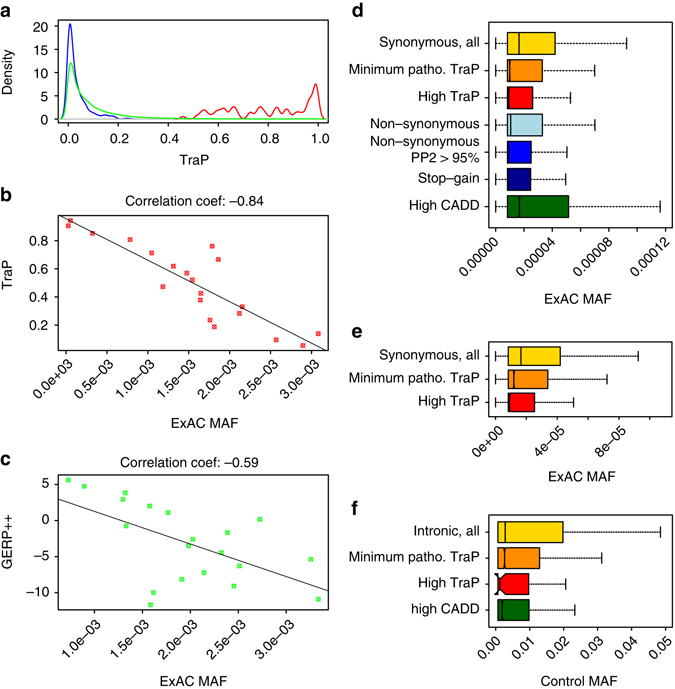
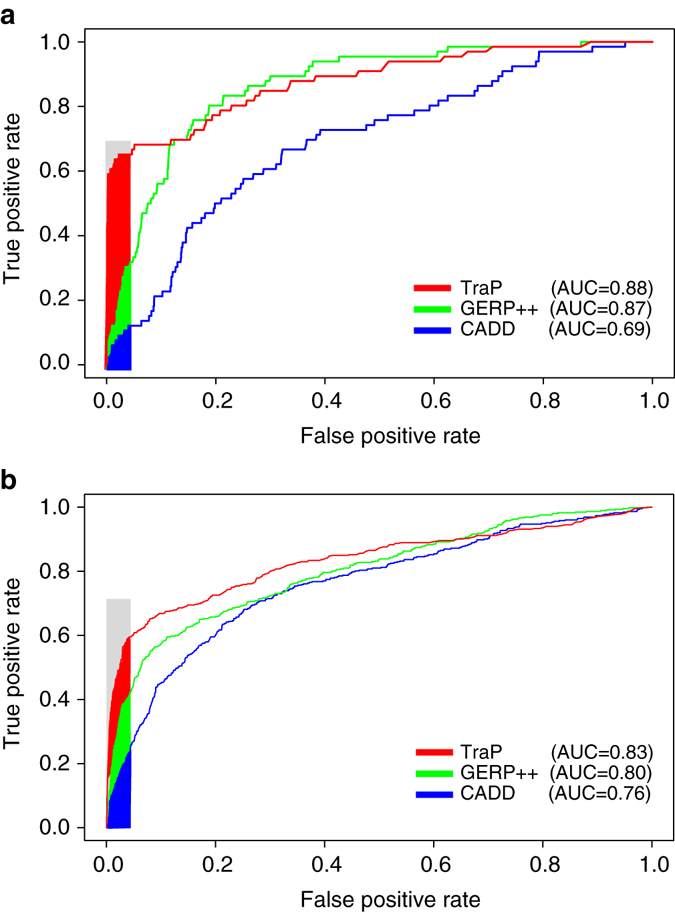
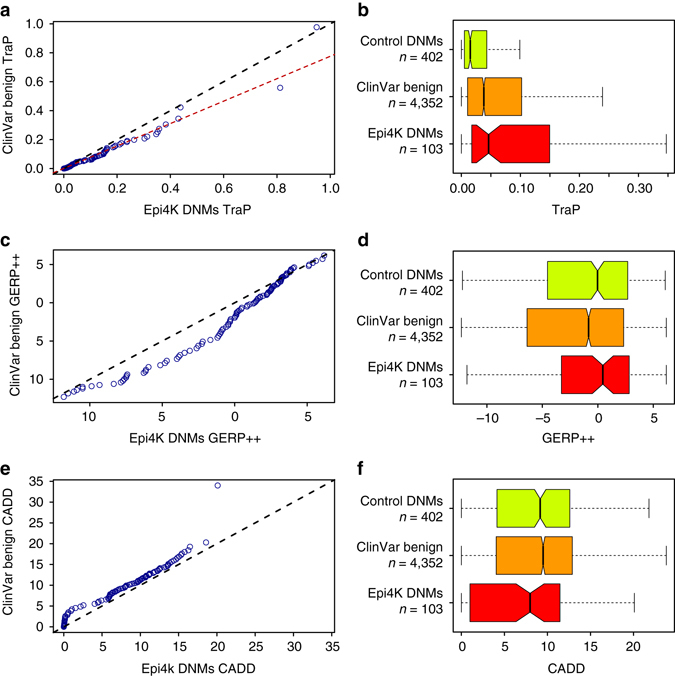
Identifying the underlying causes of disease requires accurate interpretation of genetic variants. Current methods ineffectively capture pathogenic non-coding variants in genic regions, resulting in overlooking synonymous and intronic variants when searching for disease risk. Here we present the Transcript-inferred Pathogenicity (TraP) score, which uses sequence context alterations to reliably identify non-coding variation that causes disease. High TraP scores single out extremely rare variants with lower minor allele frequencies than missense variants. TraP accurately distinguishes known pathogenic and benign variants in synonymous (AUC = 0.88) and intronic (AUC = 0.83) public datasets, dismissing benign variants with exceptionally high specificity. TraP analysis of 843 exomes from epilepsy family trios identifies synonymous variants in known epilepsy genes, thus pinpointing risk factors of disease from non-coding sequence data. TraP outperforms leading methods in identifying non-coding variants that are pathogenic and is therefore a valuable tool for use in gene discovery and the interpretation of personal genomes.While non-coding synonymous and intronic variants are often not under strong selective constraint, they can be pathogenic through affecting splicing or transcription. Here, the authors develop a score that uses sequence context alterations to predict pathogenicity of synonymous and non-coding genetic variants, and provide a web server of pre-computed scores.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Grants and funding
- P30 AG028377/AG/NIA NIH HHS/United States
- U01 NS077274/NS/NINDS NIH HHS/United States
- T32 GM007754/GM/NIGMS NIH HHS/United States
- K01 MH098126/MH/NIMH NIH HHS/United States
- R56 AI098588/AI/NIAID NIH HHS/United States
- R01 MH097993/MH/NIMH NIH HHS/United States
- U01 NS077275/NS/NINDS NIH HHS/United States
- U01 NS077367/NS/NINDS NIH HHS/United States
- RC2 MH089915/MH/NIMH NIH HHS/United States
- U01 HG007672/HG/NHGRI NIH HHS/United States
- U01 NS053998/NS/NINDS NIH HHS/United States
- U01 NS077364/NS/NINDS NIH HHS/United States
- R25 GM086262/GM/NIGMS NIH HHS/United States
- U01 MH105670/MH/NIMH NIH HHS/United States
- U01 NS077303/NS/NINDS NIH HHS/United States
- U19 AI067854/AI/NIAID NIH HHS/United States
- U01 NS077276/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
