

Pathogenesis of Lafora Disease: Transition of Soluble Glycogen to Insoluble Polyglucosan
- PMID: 28800070
- PMCID: PMC5578133
- DOI: 10.3390/ijms18081743
Pathogenesis of Lafora Disease: Transition of Soluble Glycogen to Insoluble Polyglucosan
Abstract
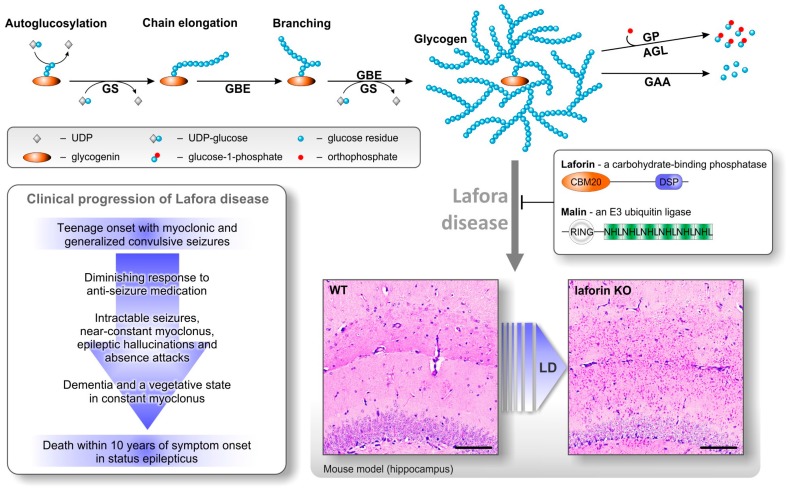
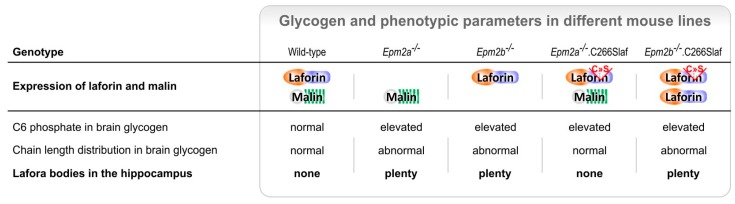
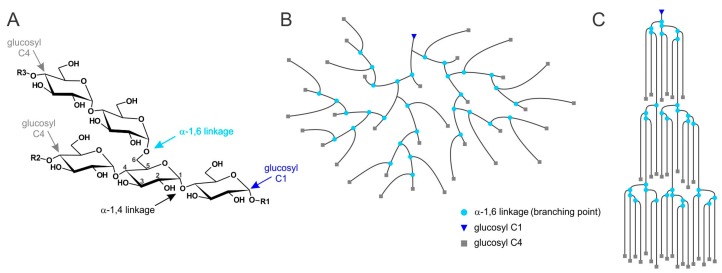
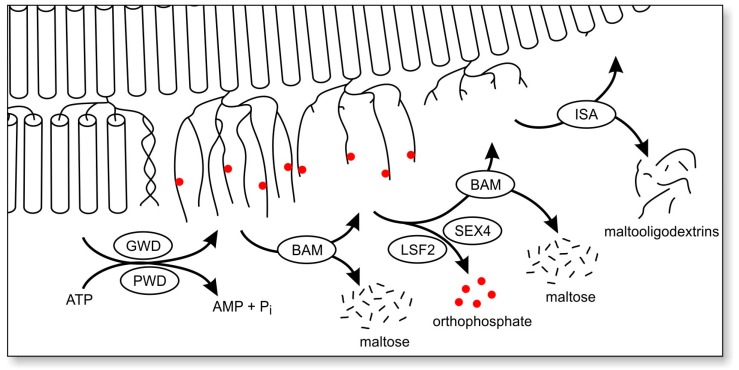
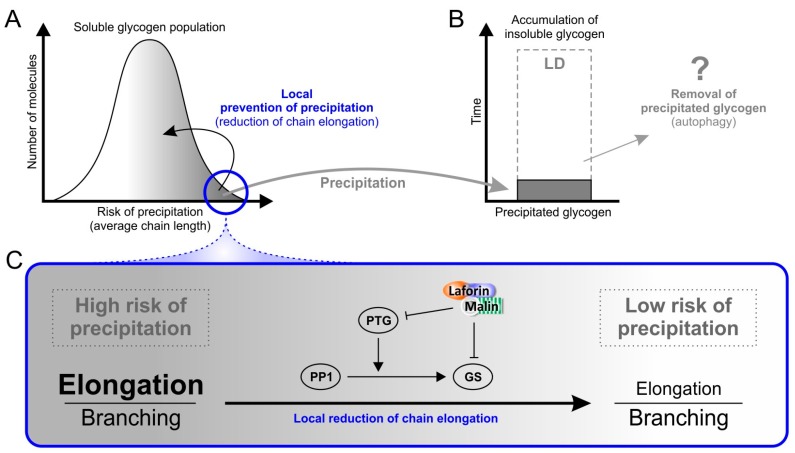
Lafora disease (LD, OMIM #254780) is a rare, recessively inherited neurodegenerative disease with adolescent onset, resulting in progressive myoclonus epilepsy which is fatal usually within ten years of symptom onset. The disease is caused by loss-of-function mutations in either of the two genes EPM2A (laforin) or EPM2B (malin). It characteristically involves the accumulation of insoluble glycogen-derived particles, named Lafora bodies (LBs), which are considered neurotoxic and causative of the disease. The pathogenesis of LD is therefore centred on the question of how insoluble LBs emerge from soluble glycogen. Recent data clearly show that an abnormal glycogen chain length distribution, but neither hyperphosphorylation nor impairment of general autophagy, strictly correlates with glycogen accumulation and the presence of LBs. This review summarizes results obtained with patients, mouse models, and cell lines and consolidates apparent paradoxes in the LD literature. Based on the growing body of evidence, it proposes that LD is predominantly caused by an impairment in chain-length regulation affecting only a small proportion of the cellular glycogen. A better grasp of LD pathogenesis will further develop our understanding of glycogen metabolism and structure. It will also facilitate the development of clinical interventions that appropriately target the underlying cause of LD.
Keywords: chain length distribution; glycogen phosphorylation; lafora disease; laforin; malin; polyglucosan body.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Serratosa J.M., Gomez-Garre P., Gallardo M.E., Anta B., de Bernabe D.B., Lindhout D., Augustijn P.B., Tassinari C.A., Malafosse R.M., Topcu M., et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the lafora type (EPM2) Hum. Mol. Genet. 1999;8:345–352. doi: 10.1093/hmg/8.2.345. - DOI - PubMed
-
- Chan E.M., Bulman D.E., Paterson A.D., Turnbull J., Andermann E., Andermann F., Rouleau G.A., Delgado-Escueta A.V., Scherer S.W., Minassian B.A. Genetic mapping of a new lafora progressive myoclonus epilepsy locus (EPM2B) on 6p22. J. Med. Genet. 2003;40:671–675. doi: 10.1136/jmg.40.9.671. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
