

The Inherent Asymmetry of DNA Replication
- PMID: 28800257
- PMCID: PMC5695668
- DOI: 10.1146/annurev-cellbio-100616-060447
The Inherent Asymmetry of DNA Replication
Abstract
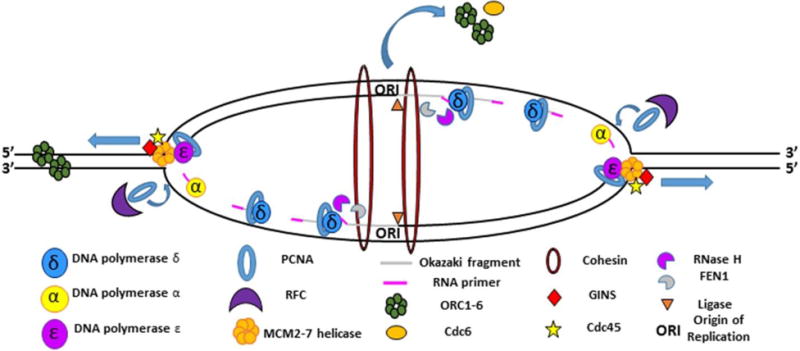
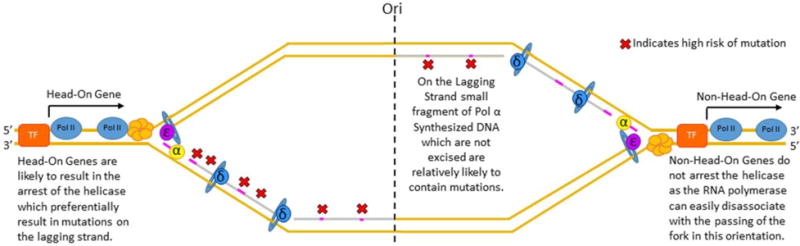
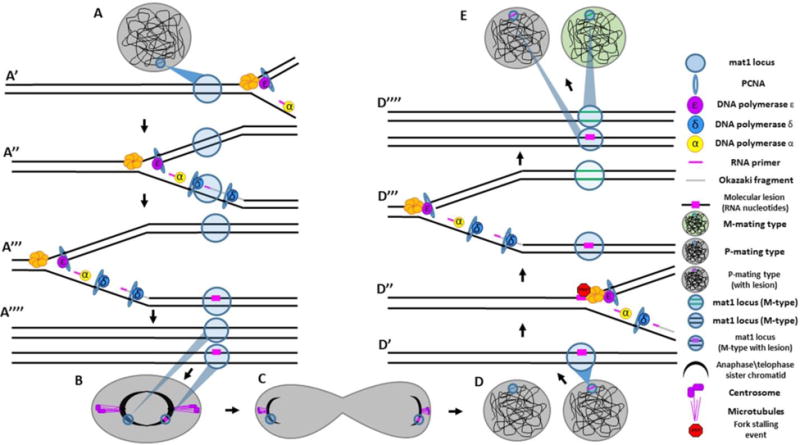
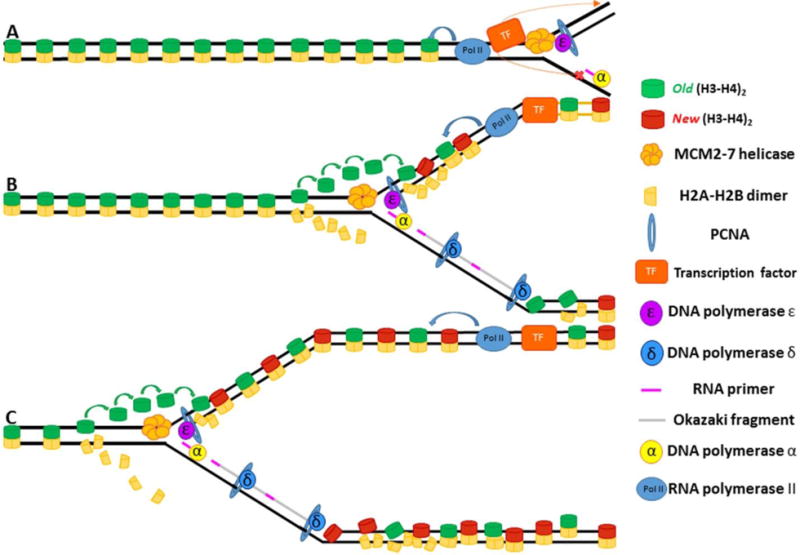
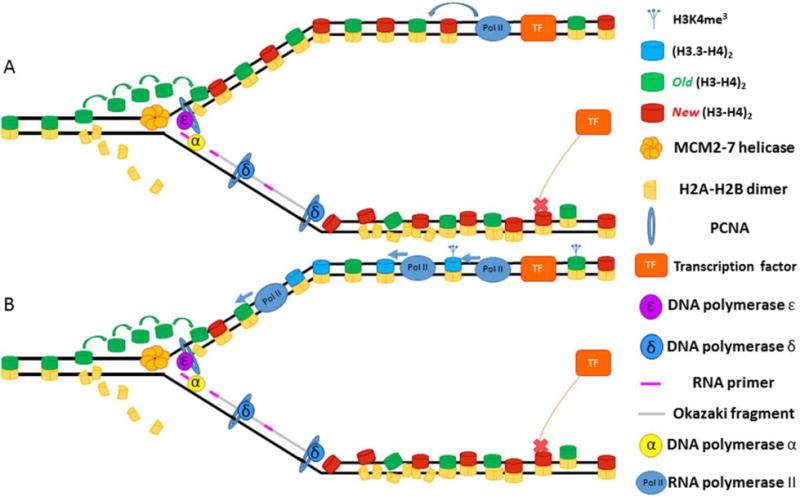
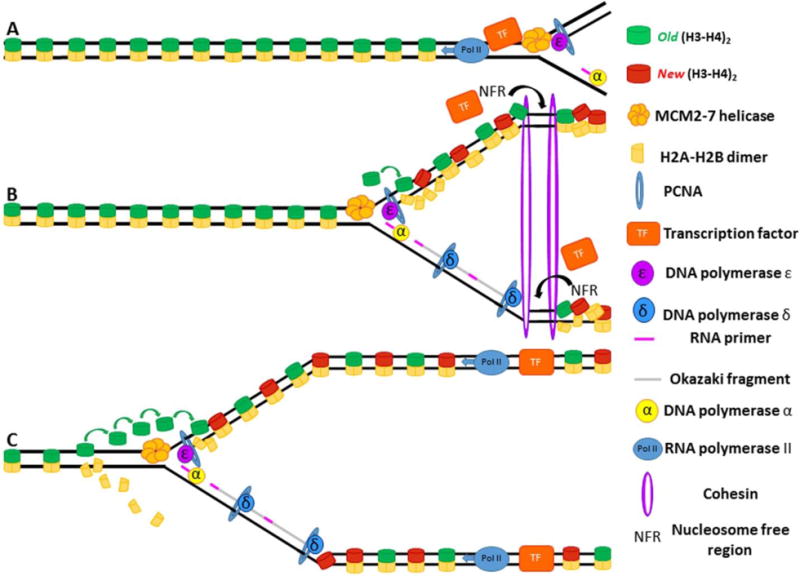
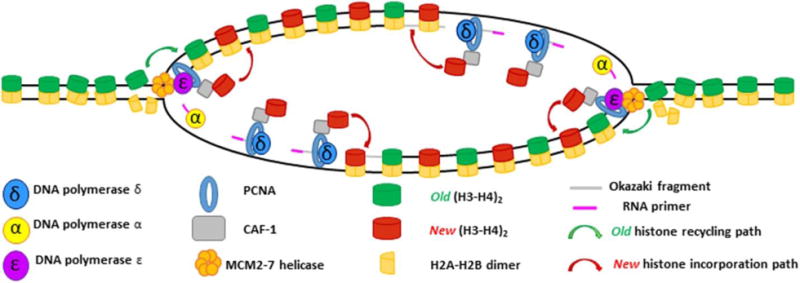
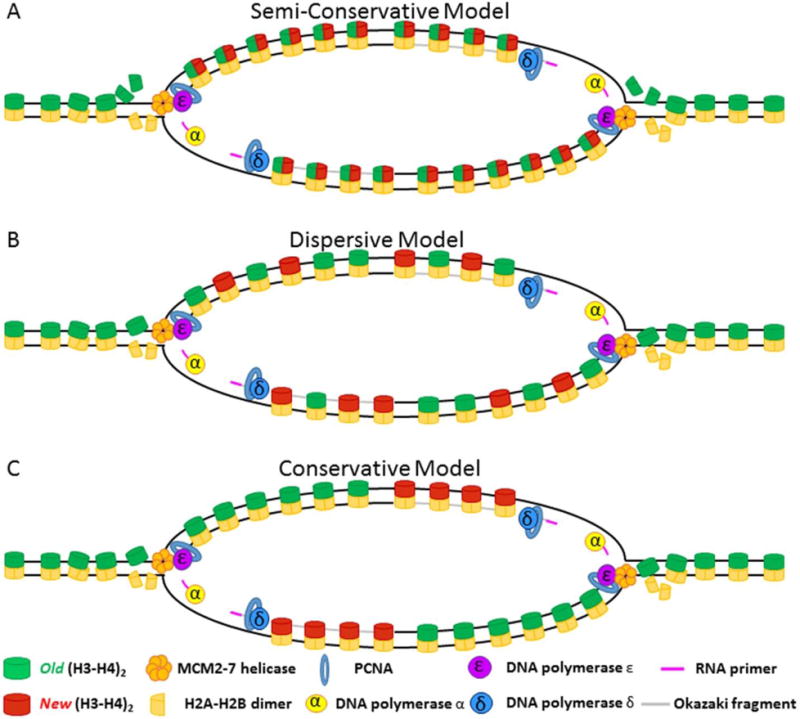
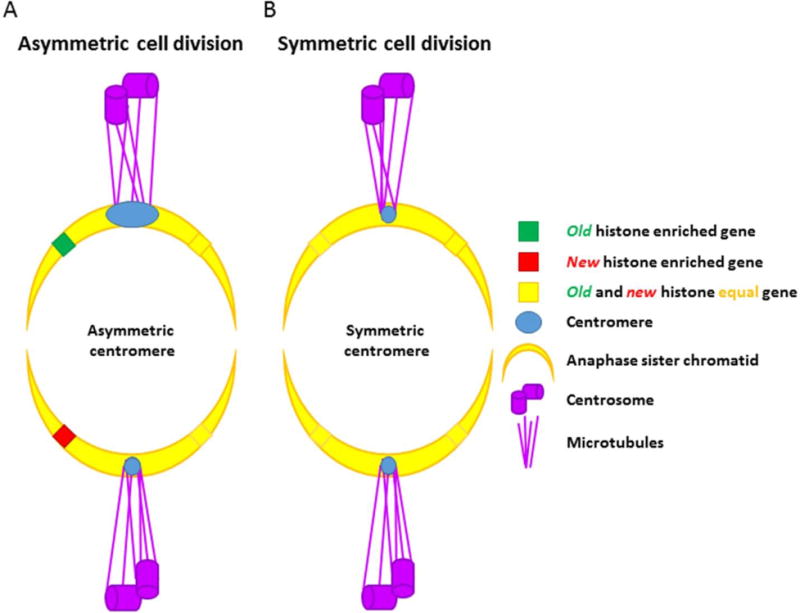
Semiconservative DNA replication has provided an elegant solution to the fundamental problem of how life is able to proliferate in a way that allows cells, organisms, and populations to survive and replicate many times over. Somewhat lost, however, in our admiration for this mechanism is an appreciation for the asymmetries that occur in the process of DNA replication. As we discuss in this review, these asymmetries arise as a consequence of the structure of the DNA molecule and the enzymatic mechanism of DNA synthesis. Increasing evidence suggests that asymmetries in DNA replication are able to play a central role in the processes of adaptation and evolution by shaping the mutagenic landscape of cells. Additionally, in eukaryotes, recent work has demonstrated that the inherent asymmetries in DNA replication may play an important role in the process of chromatin replication. As chromatin plays an essential role in defining cell identity, asymmetries generated during the process of DNA replication may play critical roles in cell fate decisions related to patterning and development.
Keywords: DNA replication; asymmetric; chromatin; epigenetic inheritance; histones.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
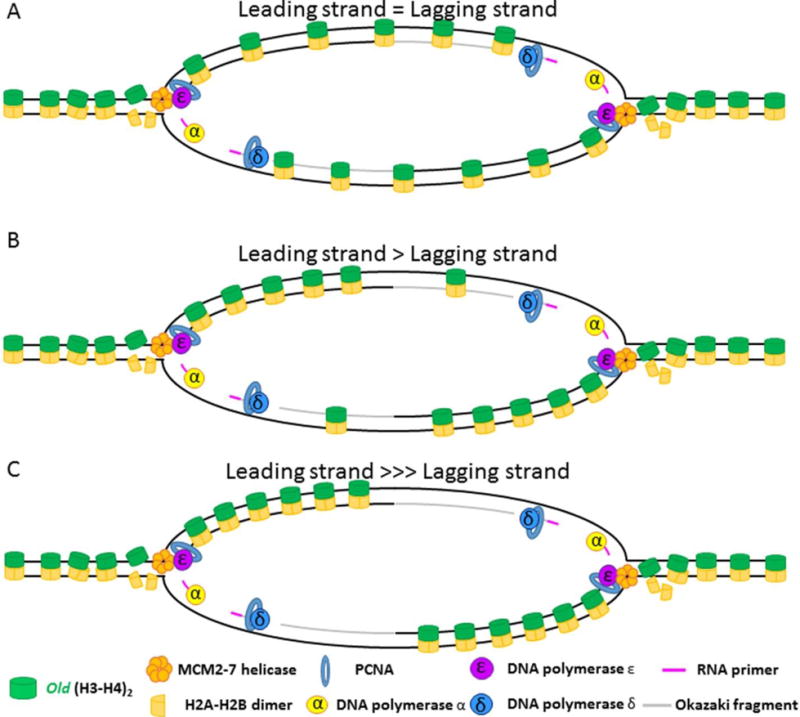
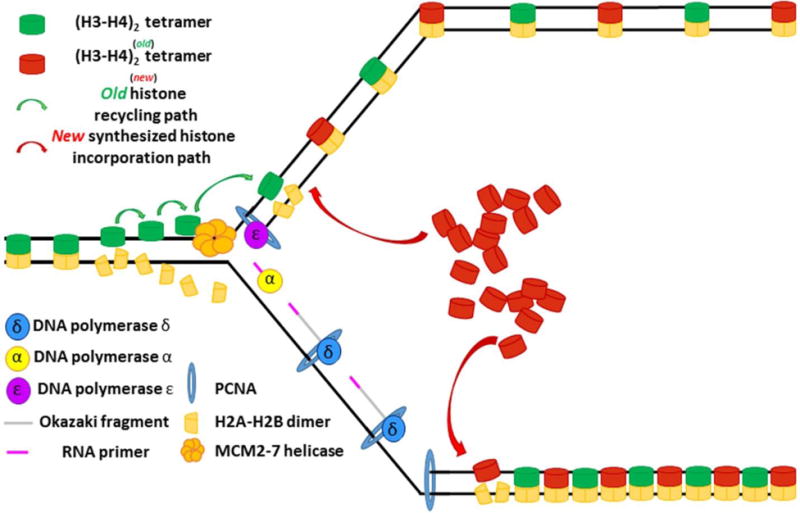
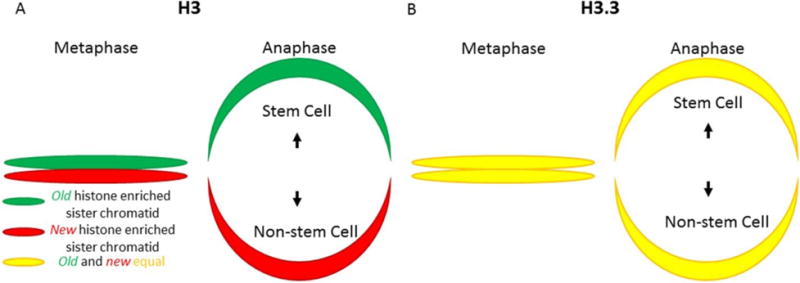
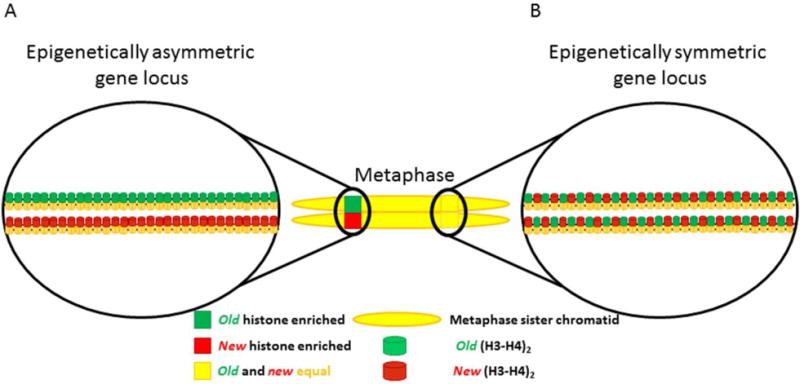
References
-
- AHMAD K, HENIKOFF S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell. 2002;9:1191–200. - PubMed
-
- ALABERT C, GROTH A. Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol. 2012;13:153–67. - PubMed
-
- ALLIS CD, JENUWEIN T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
