

Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes
- PMID: 28801211
- PMCID: PMC5803469
- DOI: 10.1016/j.joca.2017.07.020
Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes
Abstract
Objective: Mitochondrial dysfunction, oxidative stress and chondrocyte death are important contributors to the development and pathogenesis of osteoarthritis (OA). In this study, we determined the expression and role of Parkin in the clearance of damaged/dysfunctional mitochondria, regulation of reactive oxygen species (ROS) levels and chondrocyte survival under pathological conditions.
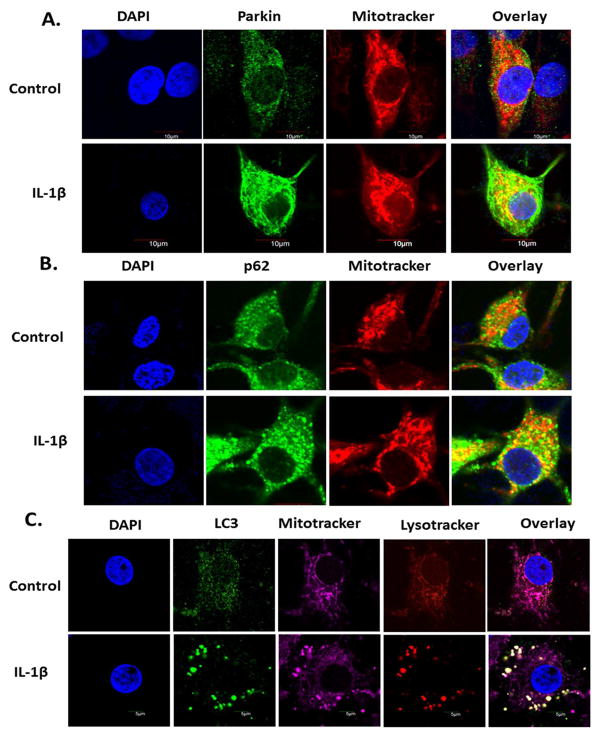
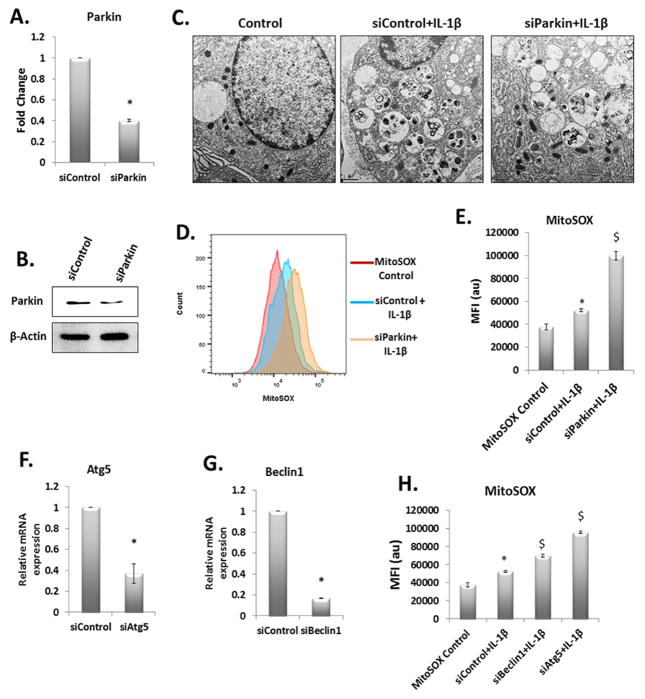
Methods: Human chondrocytes were from the unaffected area of knee OA cartilage (n = 12) and were stimulated with IL-1β to mimic pathological conditions. Mitochondrial membrane depolarization and ROS levels were determined using specific dyes and flow cytometry. Autophagy was determined by Western blotting for ATG5, Beclin1, immunofluorescence staining and confocal microscopy. Gene expression was determined by RT-qPCR. siRNA, wild-type and mutant Parkin plasmids were transfected using Amaxa system. Apoptosis was determined by PI staining of chondrocytes and TUNEL assay.
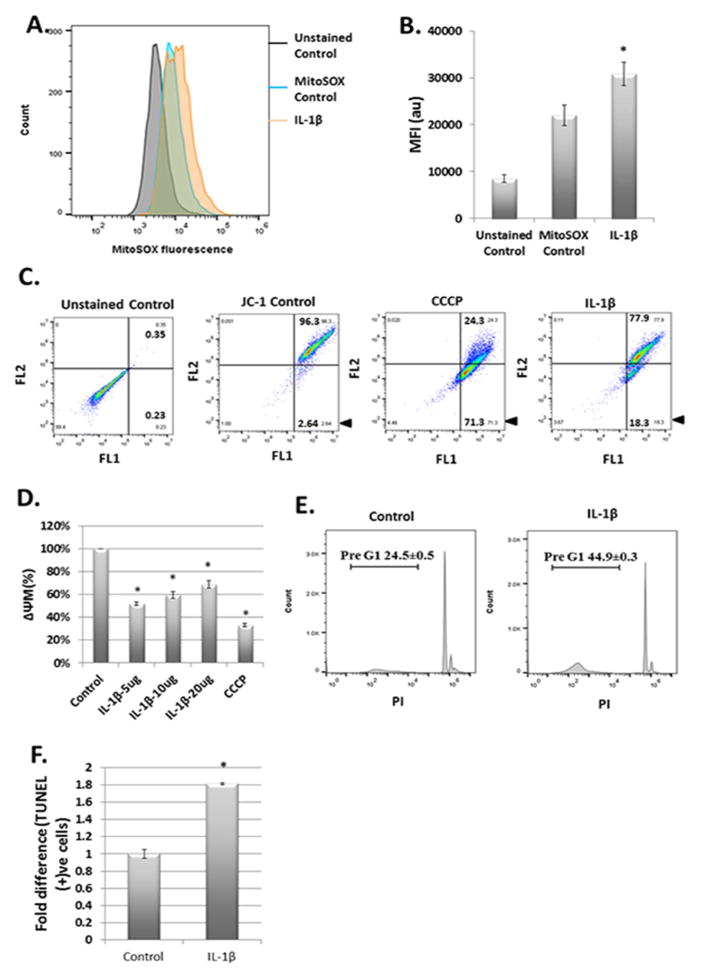
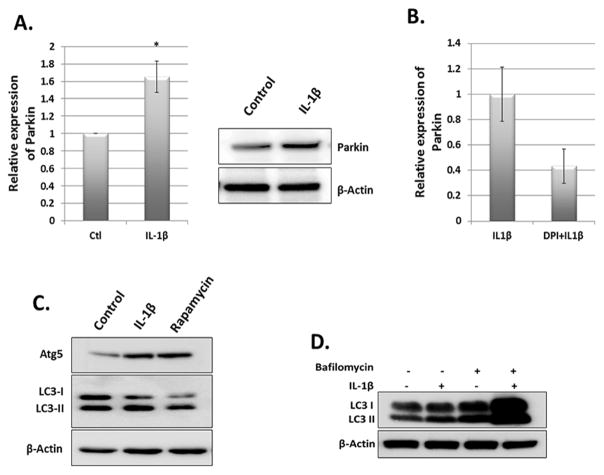
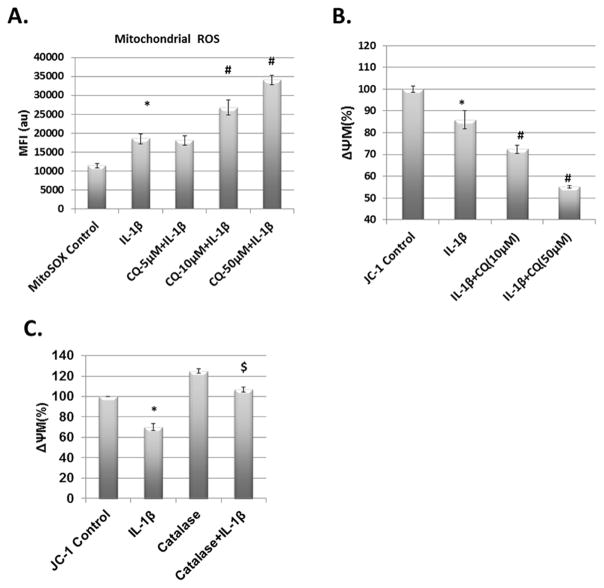
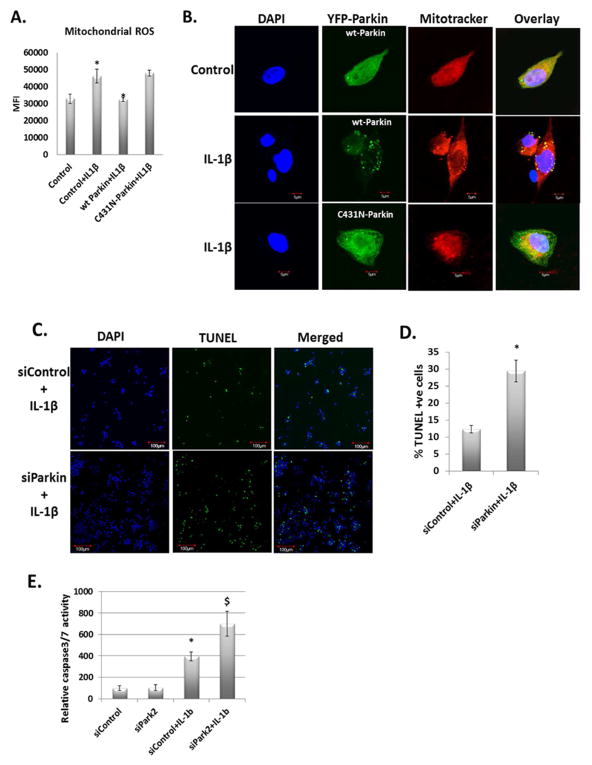
Results: IL-1β-stimulated OA chondrocytes showed high levels of ROS generation, mitochondrial membrane damage, accumulation of damaged mitochondria and higher incidence of apoptosis. IL-1β stimulation of chondrocytes with depleted Parkin expression resulted in sustained high levels of ROS, accumulation of damaged/dysfunctional mitochondria and enhanced apoptosis. Parkin translocation to depolarized/damaged mitochondria and recruitment of p62/SQSTM1 was required for the elimination of damaged/dysfunctional mitochondria in IL-1β-stimulated OA chondrocytes. Importantly we demonstrate that Parkin elimination of depolarized/damaged mitochondria required the Parkin ubiquitin ligase activity and resulted in reduced ROS levels and inhibition of apoptosis in OA chondrocytes under pathological conditions.
Conclusions: Our data demonstrates that Parkin functions to eliminate depolarized/damaged mitochondria in chondrocytes which is necessary for mitochondrial quality control, regulation of ROS levels and chondrocyte survival under pathological conditions.
Keywords: Chondrocytes; Mitochondrial dysfunction; Osteoarthritis; Parkin; ROS.
Copyright © 2017 Osteoarthritis Research Society International. Published by Elsevier Ltd. All rights reserved.
Conflict of interest statement
The authors have no conflict of interest.
Figures
Comment in
-
Mitochondria and mitophagy: biosensors for cartilage degradation and osteoarthritis.Osteoarthritis Cartilage. 2018 Aug;26(8):989-991. doi: 10.1016/j.joca.2018.05.018. Epub 2018 May 29. Osteoarthritis Cartilage. 2018. PMID: 29857157 No abstract available.
References
-
- Glyn-Jones S, Palmer AJ, Agricola R, Price AJ, Vincent TL, Weinans H, et al. Osteoarthritis. Lancet. 2015;386:376–387. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
