

Validation of a Targeted RNA Sequencing Assay for Kinase Fusion Detection in Solid Tumors
- PMID: 28802831
- PMCID: PMC5975628
- DOI: 10.1016/j.jmoldx.2017.05.006
Validation of a Targeted RNA Sequencing Assay for Kinase Fusion Detection in Solid Tumors
Abstract
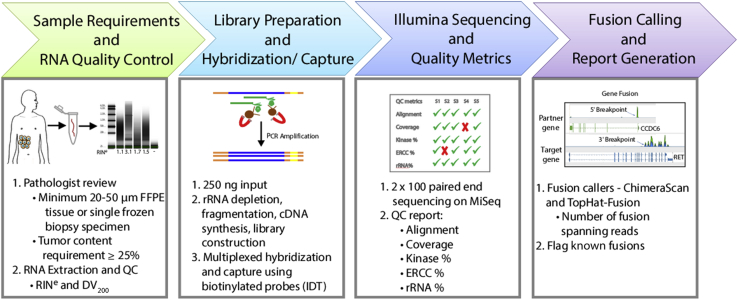
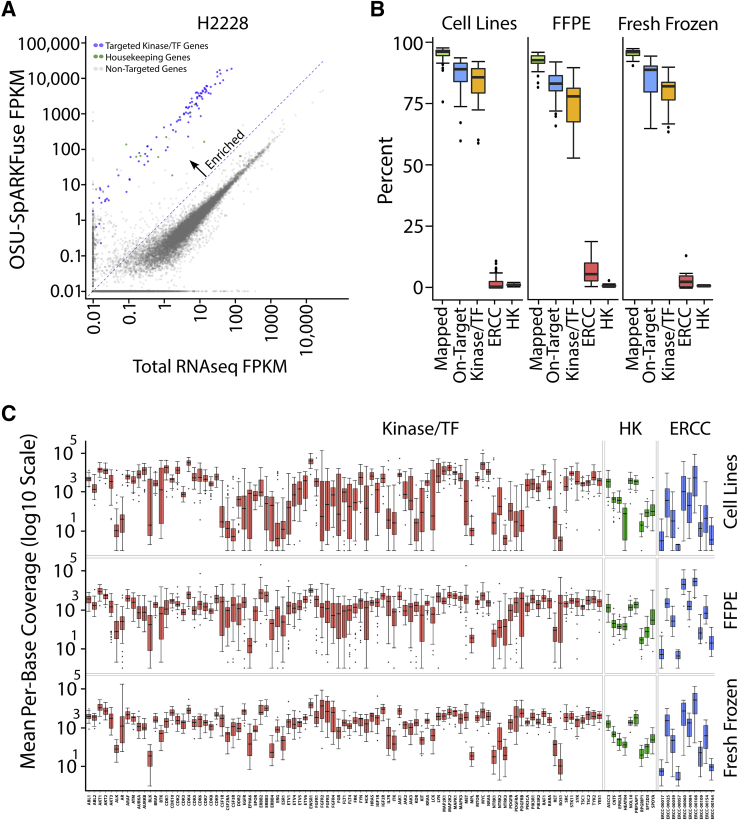
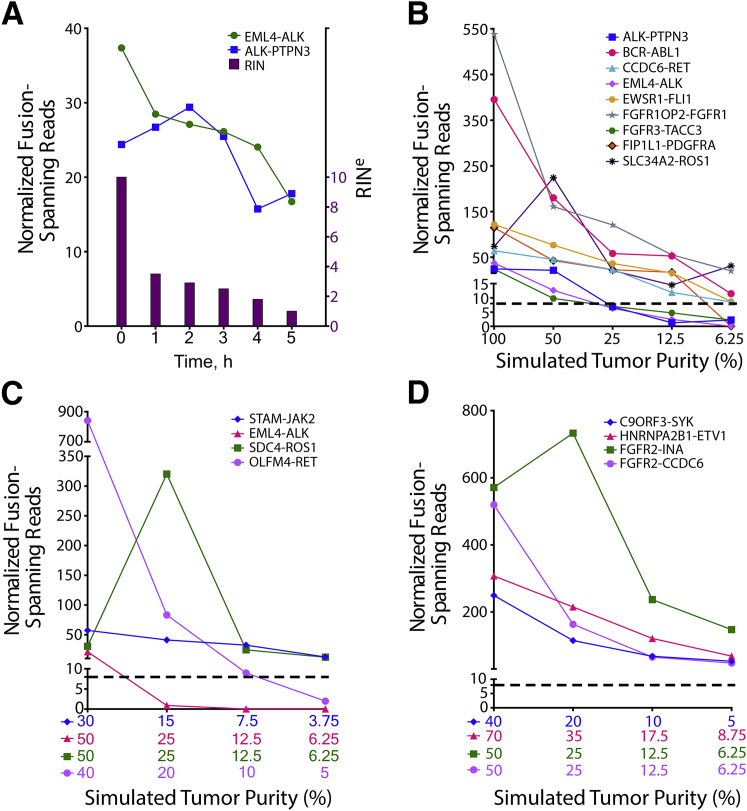
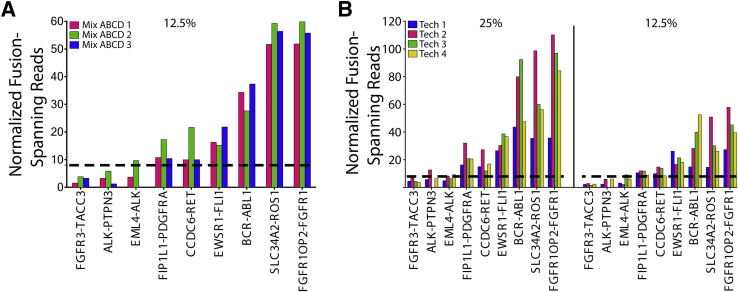
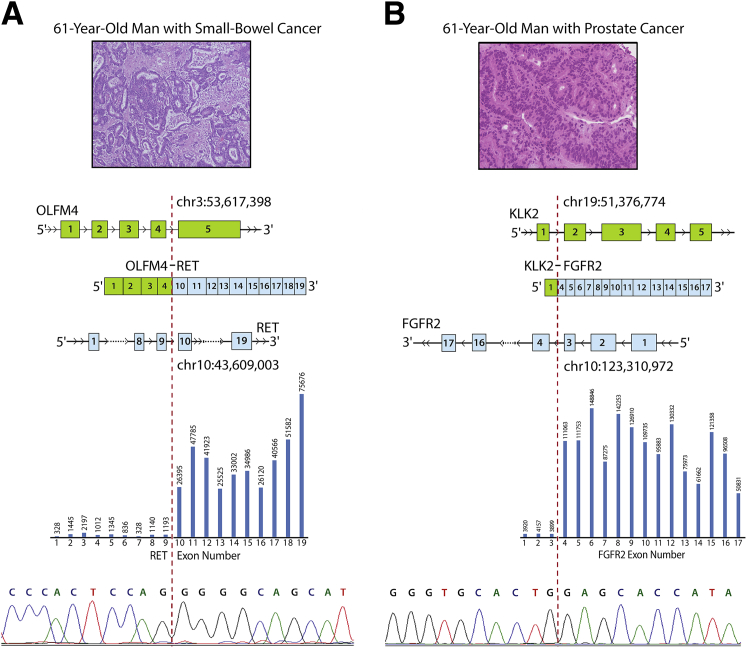
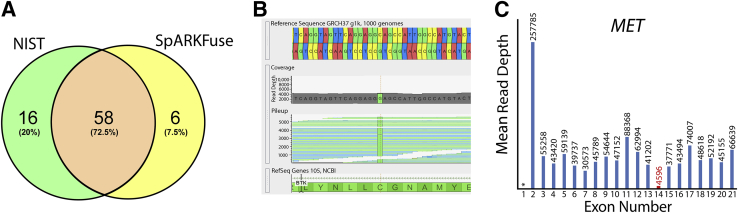
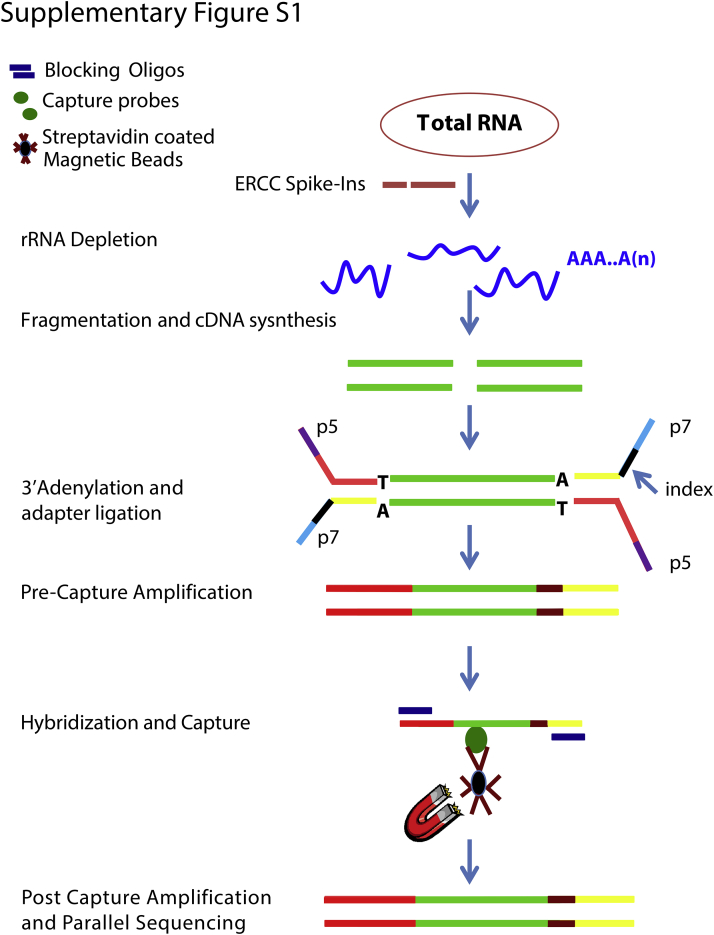
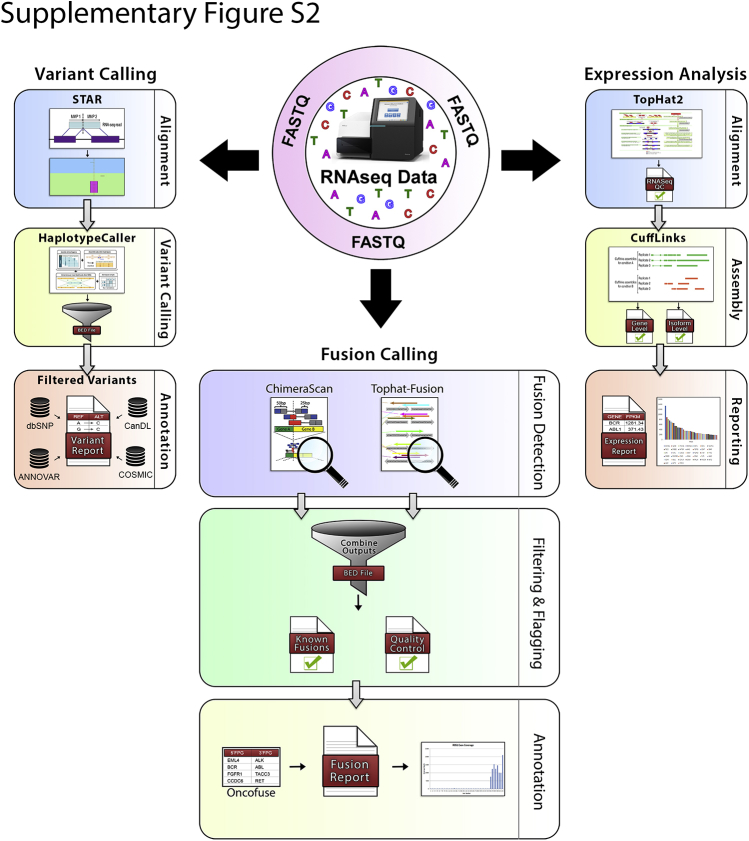
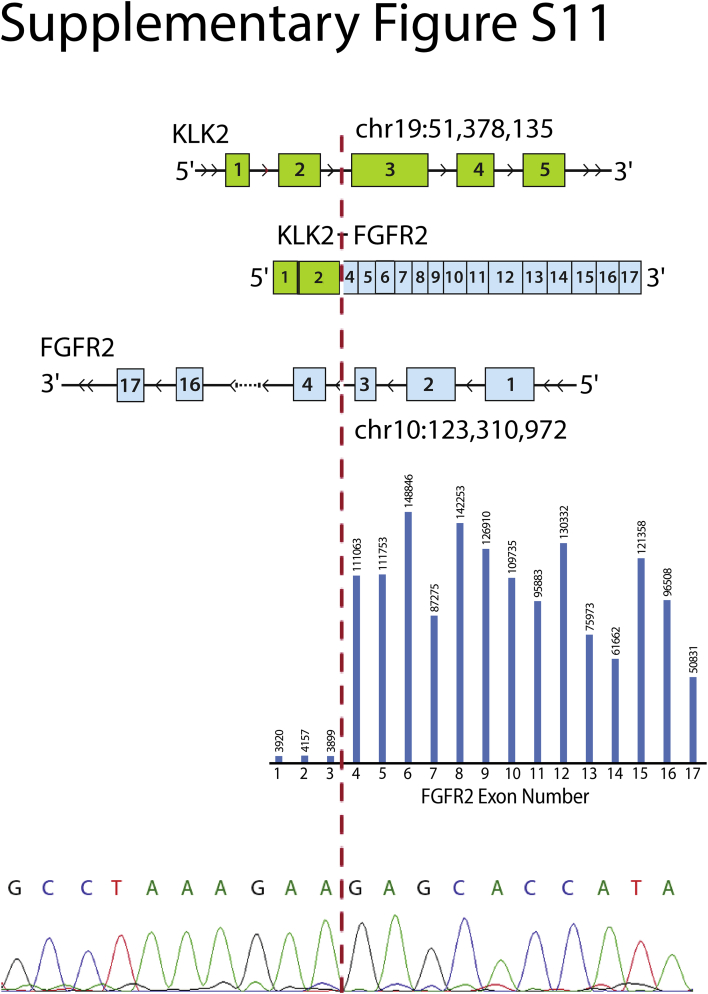
Kinase gene fusions are important drivers of oncogenic transformation and can be inhibited with targeted therapies. Clinical grade diagnostics using RNA sequencing to detect gene rearrangements in solid tumors are limited, and the few that are available require prior knowledge of fusion break points. To address this, we have analytically validated a targeted RNA sequencing assay (OSU-SpARKFuse) for fusion detection that interrogates complete transcripts from 93 kinase and transcription factor genes. From a total of 74 positive and 36 negative control samples, OSU-SpARKFuse had 93.3% sensitivity and 100% specificity for fusion detection. Assessment of repeatability and reproducibility revealed 96.3% and 94.4% concordance between intrarun and interrun technical replicates, respectively. Application of this assay on prospective patient samples uncovered OLFM4 as a novel RET fusion partner in a small-bowel cancer and led to the discovery of a KLK2-FGFR2 fusion in a patient with prostate cancer who subsequently underwent treatment with a pan-fibroblast growth factor receptor inhibitor. Beyond fusion detection, OSU-SpARKFuse has built-in capabilities for discovery research, including gene expression analysis, detection of single-nucleotide variants, and identification of alternative splicing events.
Copyright © 2017 American Society for Investigative Pathology and the Association for Molecular Pathology. Published by Elsevier Inc. All rights reserved.
Figures
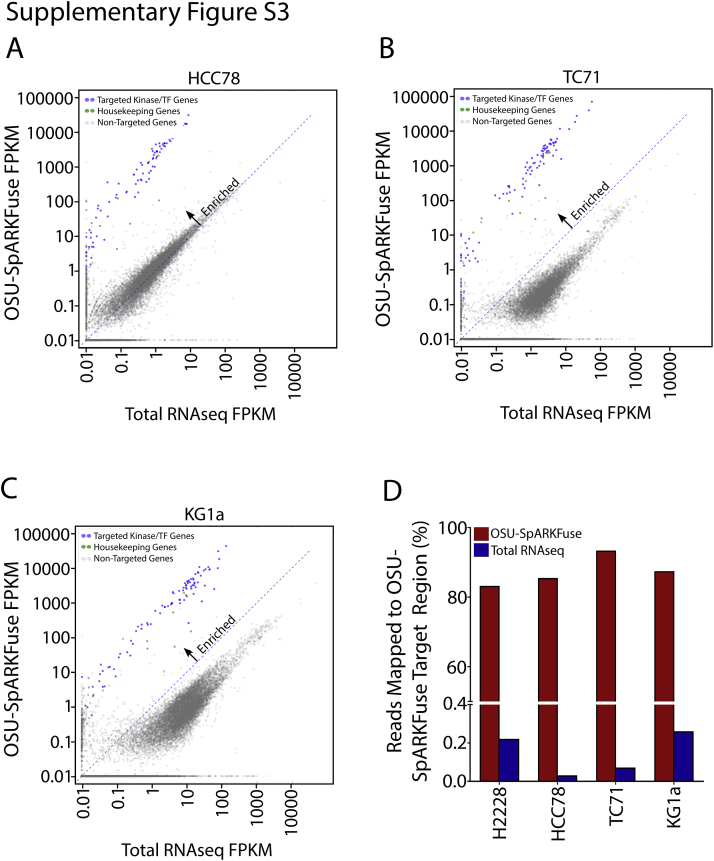
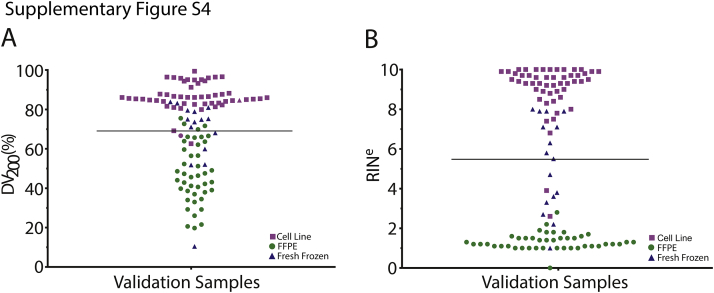
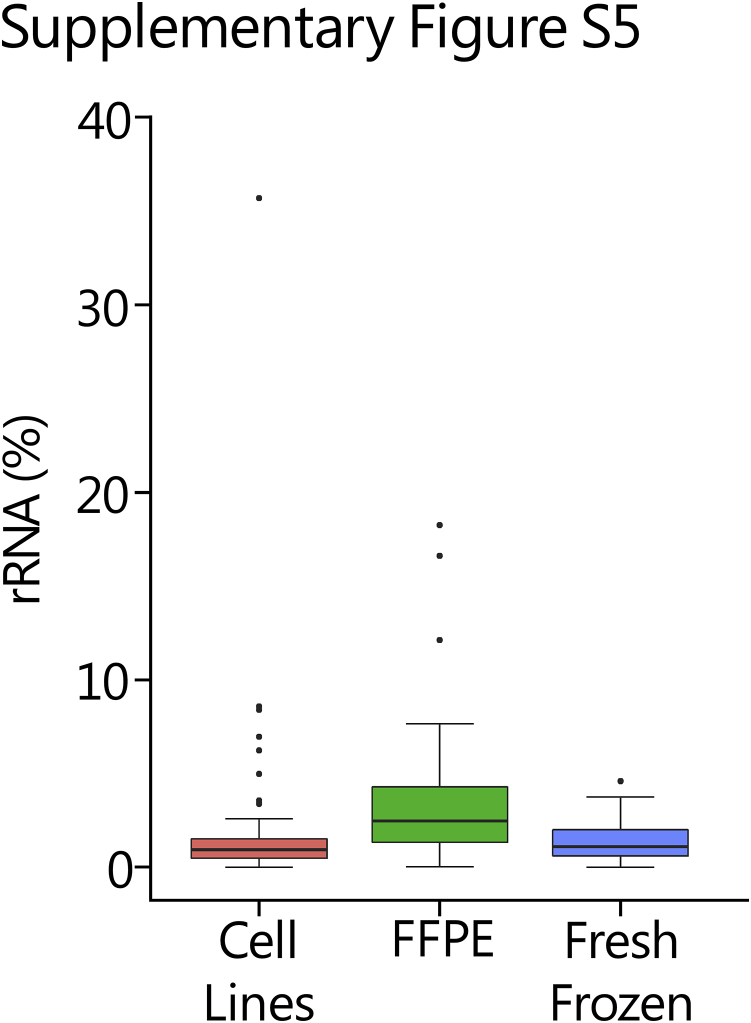
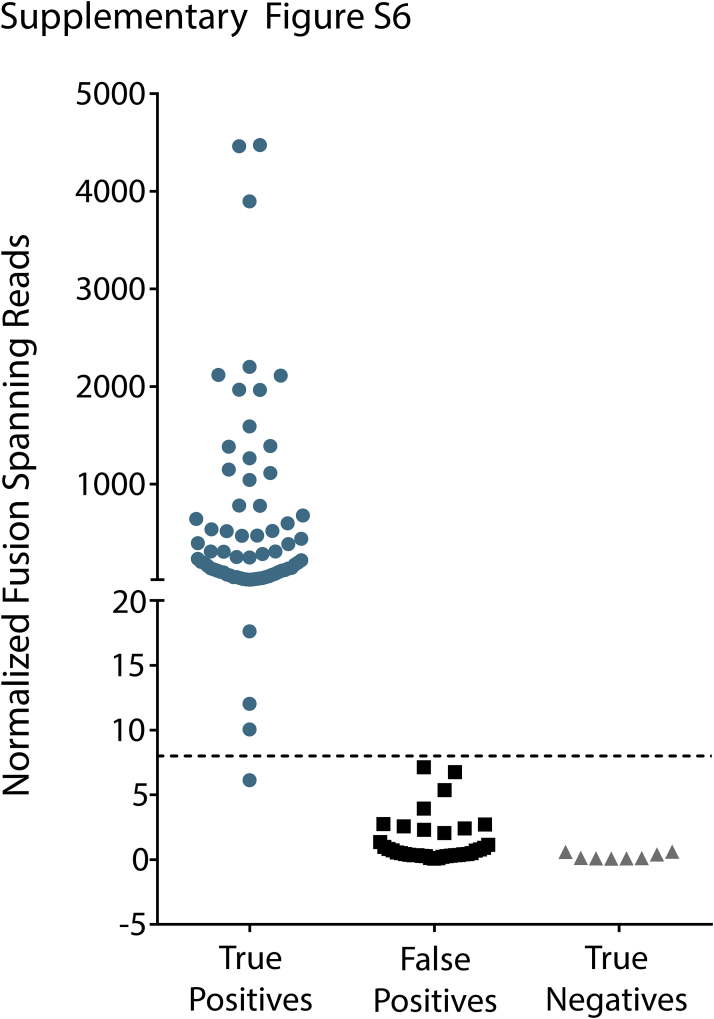
References
-
- Nowell P.C., Hungerford D.A. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst. 1960;25:85–109. - PubMed
-
- Nowell P.C., Hungerford D.A. Chromosome studies in human leukemia, II: chronic granulocytic leukemia. J Natl Cancer Inst. 1961;27:1013–1035. - PubMed
-
- Mertens F., Johansson B., Fioretos T., Mitelman F. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15:371–381. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
