

Navigating the conformational landscape of G protein-coupled receptor kinases during allosteric activation
- PMID: 28808053
- PMCID: PMC5625036
- DOI: 10.1074/jbc.M117.807461
Navigating the conformational landscape of G protein-coupled receptor kinases during allosteric activation
Abstract
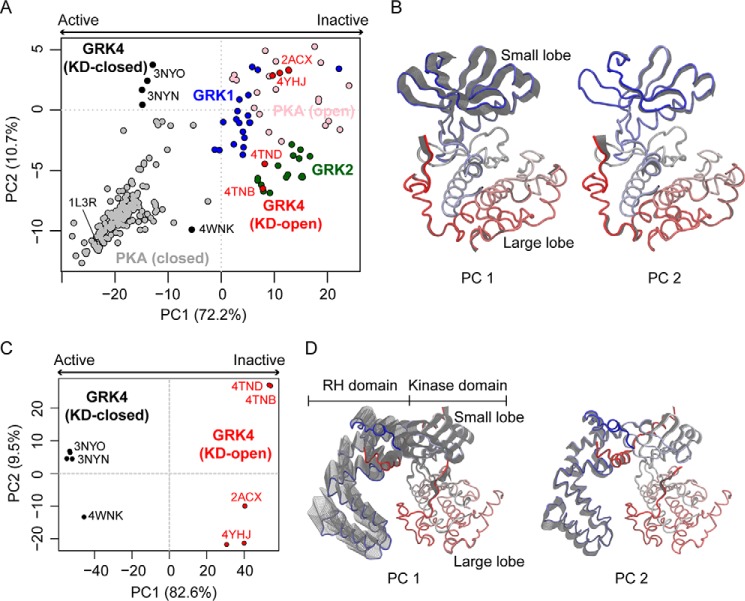
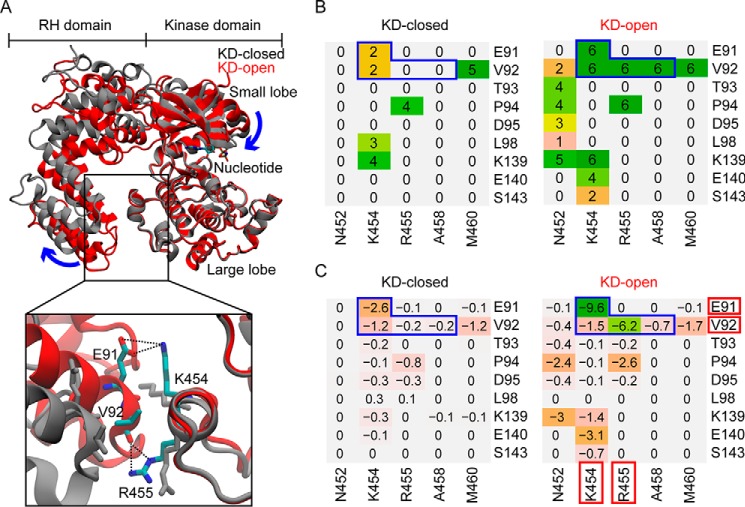
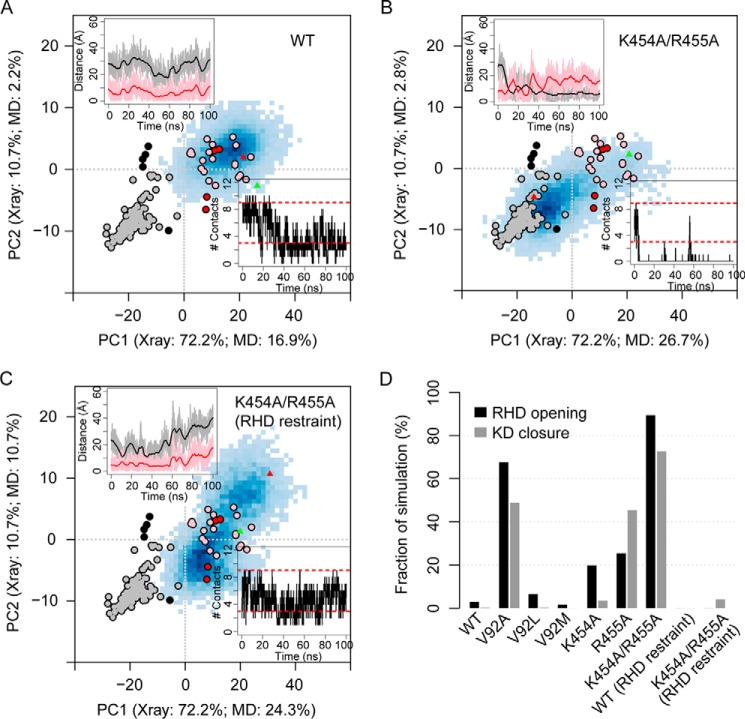
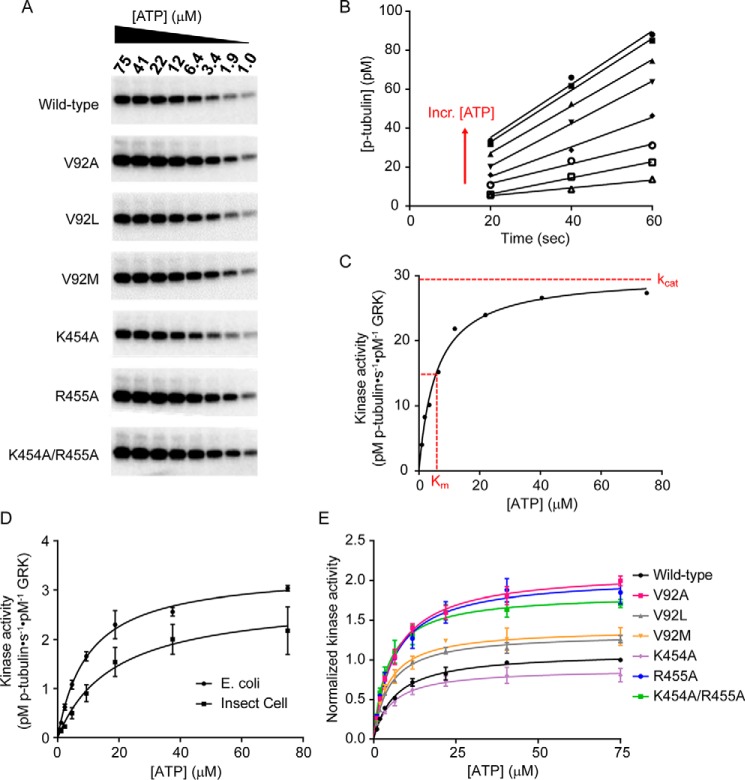
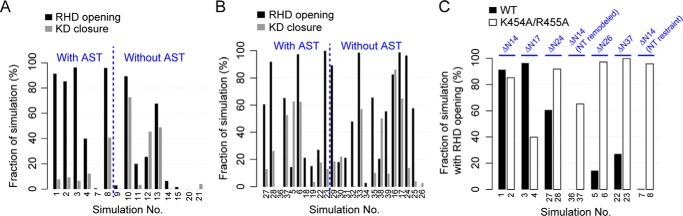
G protein-coupled receptors (GPCRs) are essential for transferring extracellular signals into carefully choreographed intracellular responses controlling diverse aspects of cell physiology. The duration of GPCR-mediated signaling is primarily regulated via GPCR kinase (GRK)-mediated phosphorylation of activated receptors. Although many GRK structures have been reported, the mechanisms underlying GRK activation are not well-understood, in part because it is unknown how these structures map to the conformational landscape available to this enzyme family. Unlike most other AGC kinases, GRKs rely on their interaction with GPCRs for activation and not phosphorylation. Here, we used principal component analysis of available GRK and protein kinase A crystal structures to identify their dominant domain motions and to provide a framework that helps evaluate how close each GRK structure is to being a catalytically competent state. Our results indicated that disruption of an interface formed between the large lobe of the kinase domain and the regulator of G protein signaling homology domain (RHD) is highly correlated with establishment of the active conformation. By introducing point mutations in the GRK5 RHD-kinase domain interface, we show with both in silico and in vitro experiments that perturbation of this interface leads to higher phosphorylation activity. Navigation of the conformational landscape defined by this bioinformatics-based study is likely common to all GPCR-activated GRKs.
Keywords: G protein-coupled receptor (GPCR); Michaelis-Menten; allosteric regulation; molecular dynamics; serine/threonine protein kinase; structure-function.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Shukla A. K., Singh G., and Ghosh E. (2014) Emerging structural insights into biased GPCR signaling. Trends Biochem. Sci. 39, 594–602 - PubMed
-
- Rockman H. A., Koch W. J., and Lefkowitz R. J. (2002) Seven-transmembrane-spanning receptors and heart function. Nature 415, 206–212 - PubMed
-
- Ferguson S. S. (2001) Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol. Rev. 53, 1–24 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
