

Genomic Evolution of Breast Cancer Metastasis and Relapse
- PMID: 28810143
- PMCID: PMC5559645
- DOI: 10.1016/j.ccell.2017.07.005
Genomic Evolution of Breast Cancer Metastasis and Relapse
Abstract
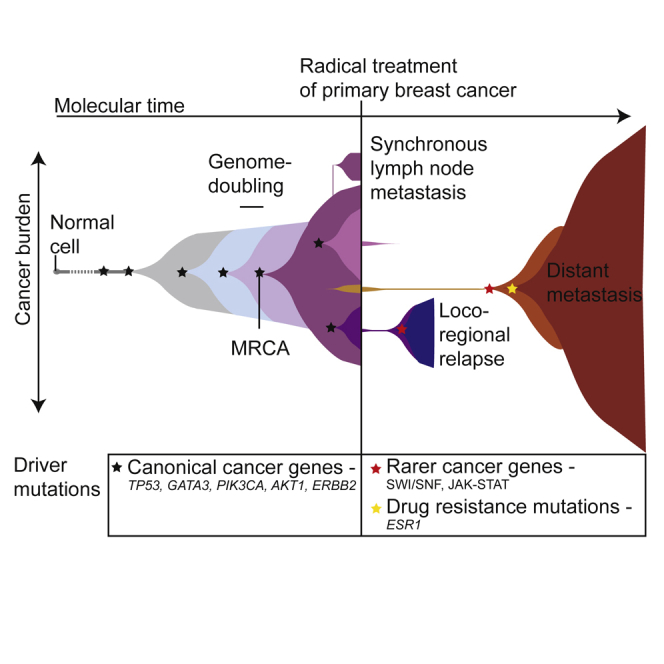
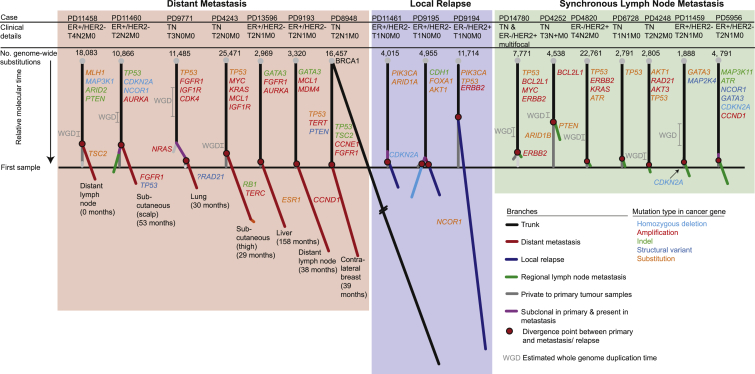
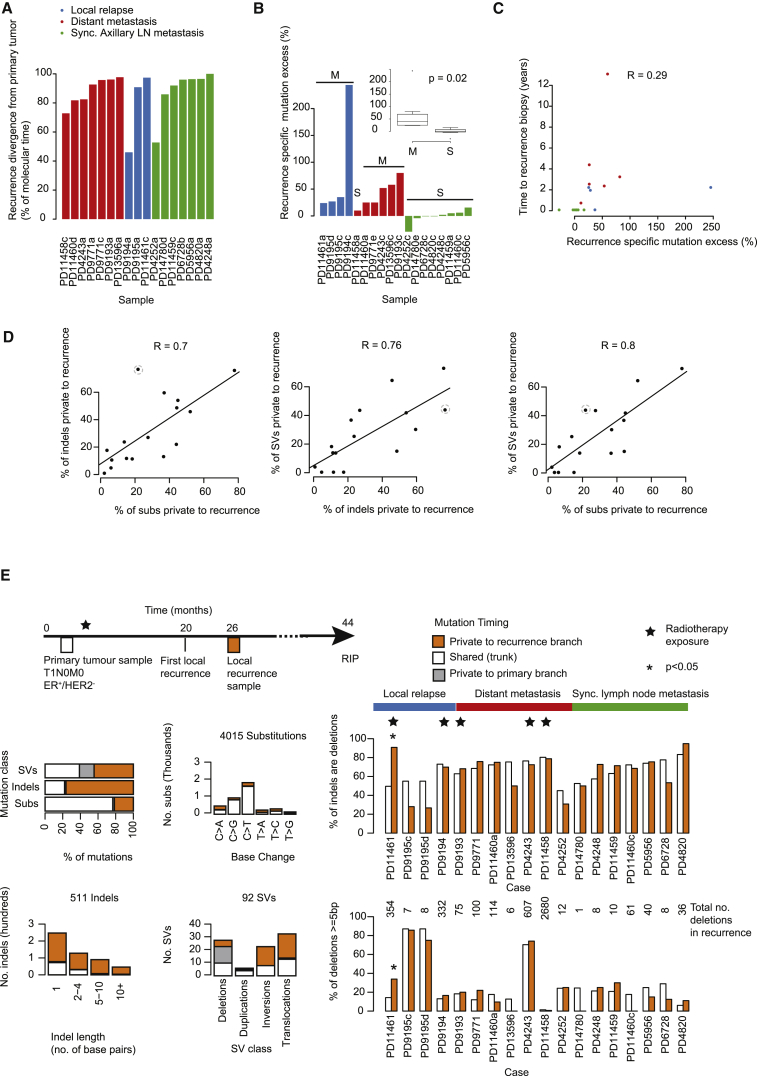
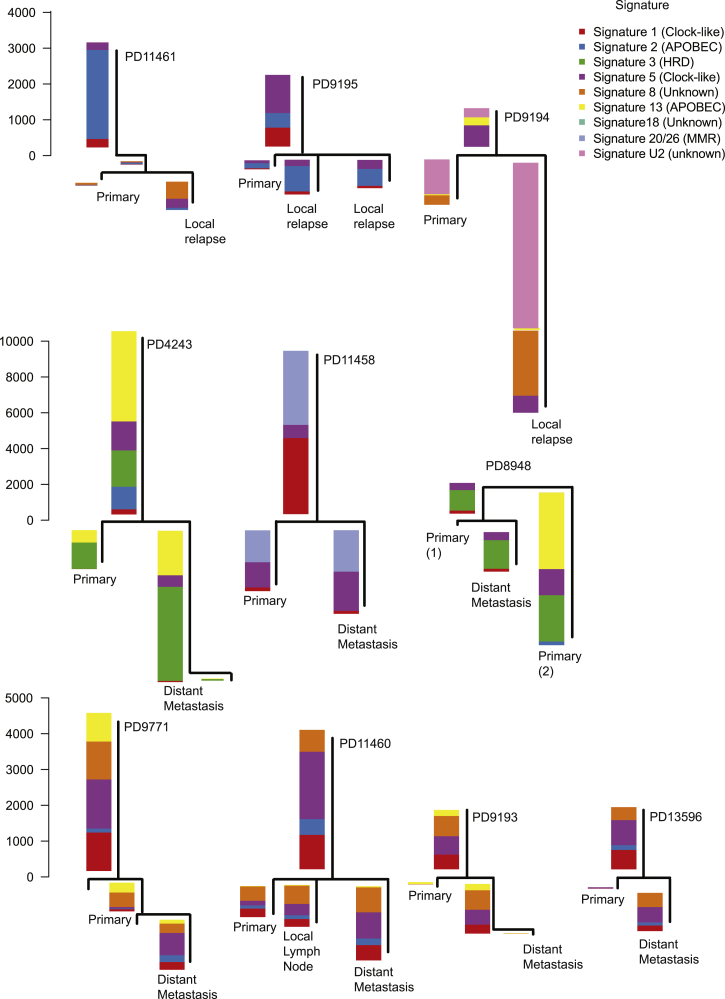
Patterns of genomic evolution between primary and metastatic breast cancer have not been studied in large numbers, despite patients with metastatic breast cancer having dismal survival. We sequenced whole genomes or a panel of 365 genes on 299 samples from 170 patients with locally relapsed or metastatic breast cancer. Several lines of analysis indicate that clones seeding metastasis or relapse disseminate late from primary tumors, but continue to acquire mutations, mostly accessing the same mutational processes active in the primary tumor. Most distant metastases acquired driver mutations not seen in the primary tumor, drawing from a wider repertoire of cancer genes than early drivers. These include a number of clinically actionable alterations and mutations inactivating SWI-SNF and JAK2-STAT3 pathways.
Keywords: breast cancer; genomics; metastasis; relapse; somatic mutation.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
New Views into the Genetic Landscape of Metastatic Breast Cancer.Cancer Cell. 2017 Aug 14;32(2):131-133. doi: 10.1016/j.ccell.2017.07.011. Cancer Cell. 2017. PMID: 28810140
References
-
- Aas T., Borresen A.L., Geisler S., Smith-Sorensen B., Johnsen H., Varhaug J.E., Akslen L.A., Lonning P.E. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat. Med. 1996;2:811–814. - PubMed
-
- Arnedos M., Vicier C., Loi S., Lefebvre C., Michiels S., Bonnefoi H., Andre F. Precision medicine for metastatic breast cancer–limitations and solutions. Nat. Rev. Clin. Oncol. 2015;12:693–704. - PubMed
-
- Balko J.M., Giltnane J.M., Wang K., Schwarz L.J., Young C.D., Cook R.S., Owens P., Sanders M.E., Kuba M.G., Sanchez V. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4:232–245. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
