

Application of a Bayesian non-linear model hybrid scheme to sequence data for genomic prediction and QTL mapping
- PMID: 28810831
- PMCID: PMC5558724
- DOI: 10.1186/s12864-017-4030-x
Application of a Bayesian non-linear model hybrid scheme to sequence data for genomic prediction and QTL mapping
Abstract
Background: Using whole genome sequence data might improve genomic prediction accuracy, when compared with high-density SNP arrays, and could lead to identification of casual mutations affecting complex traits. For some traits, the most accurate genomic predictions are achieved with non-linear Bayesian methods. However, as the number of variants and the size of the reference population increase, the computational time required to implement these Bayesian methods (typically with Monte Carlo Markov Chain sampling) becomes unfeasibly long.
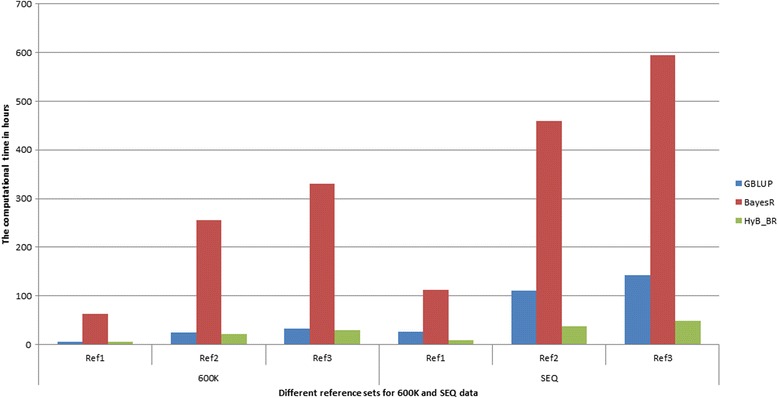
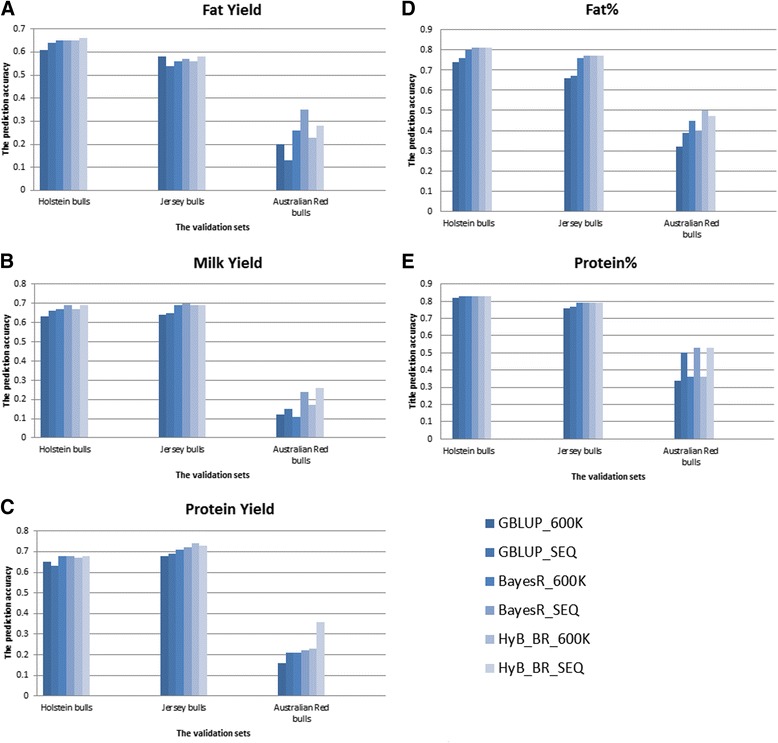
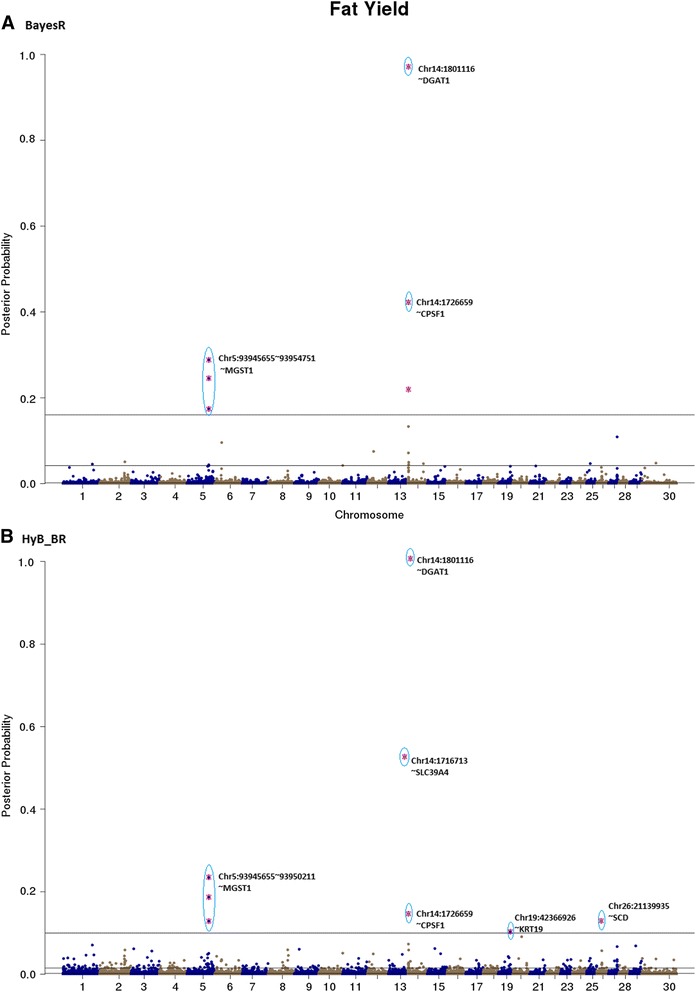
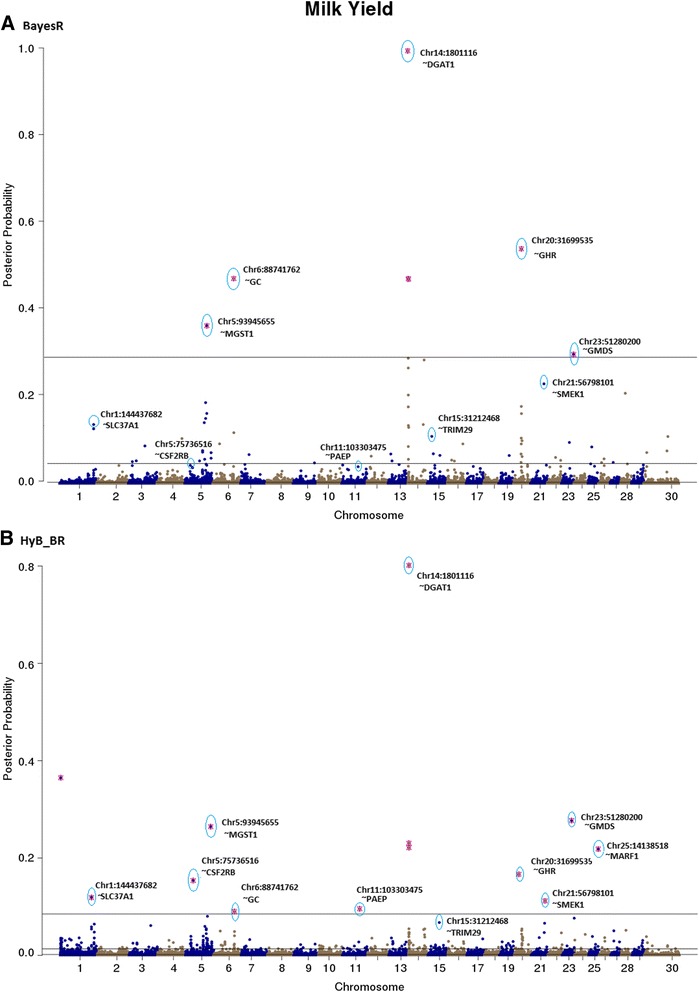
Results: Here, we applied a new method, HyB_BR (for Hybrid BayesR), which implements a mixture model of normal distributions and hybridizes an Expectation-Maximization (EM) algorithm followed by Markov Chain Monte Carlo (MCMC) sampling, to genomic prediction in a large dairy cattle population with imputed whole genome sequence data. The imputed whole genome sequence data included 994,019 variant genotypes of 16,214 Holstein and Jersey bulls and cows. Traits included fat yield, milk volume, protein kg, fat% and protein% in milk, as well as fertility and heat tolerance. HyB_BR achieved genomic prediction accuracies as high as the full MCMC implementation of BayesR, both for predicting a validation set of Holstein and Jersey bulls (multi-breed prediction) and a validation set of Australian Red bulls (across-breed prediction). HyB_BR had a ten fold reduction in compute time, compared with the MCMC implementation of BayesR (48 hours versus 594 hours). We also demonstrate that in many cases HyB_BR identified sequence variants with a high posterior probability of affecting the milk production or fertility traits that were similar to those identified in BayesR. For heat tolerance, both HyB_BR and BayesR found variants in or close to promising candidate genes associated with this trait and not detected by previous studies.
Conclusions: The results demonstrate that HyB_BR is a feasible method for simultaneous genomic prediction and QTL mapping with whole genome sequence in large reference populations.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no completing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

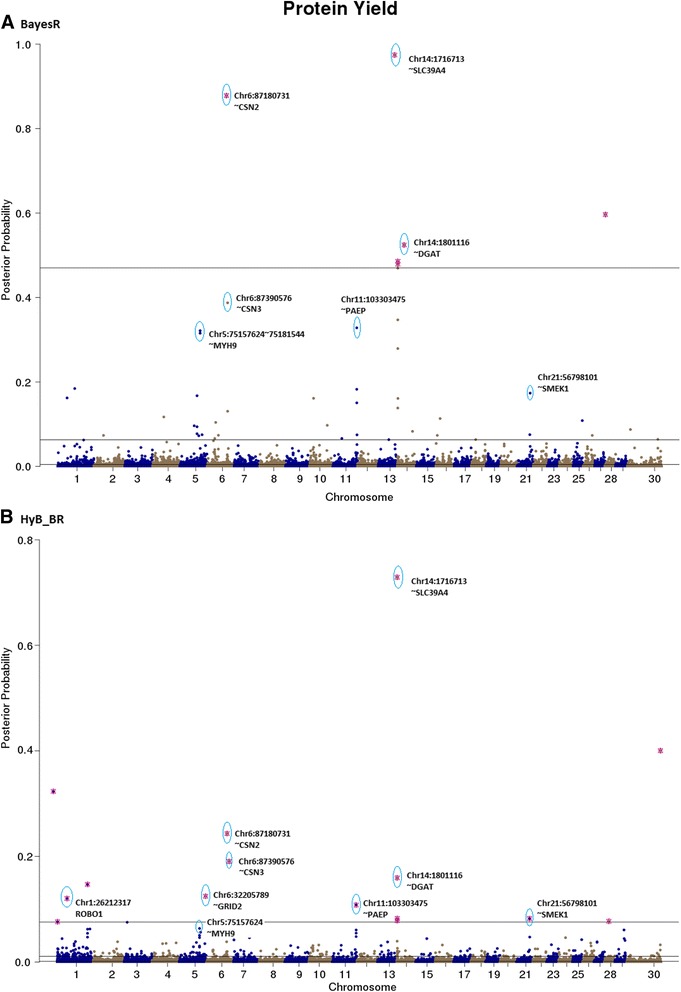
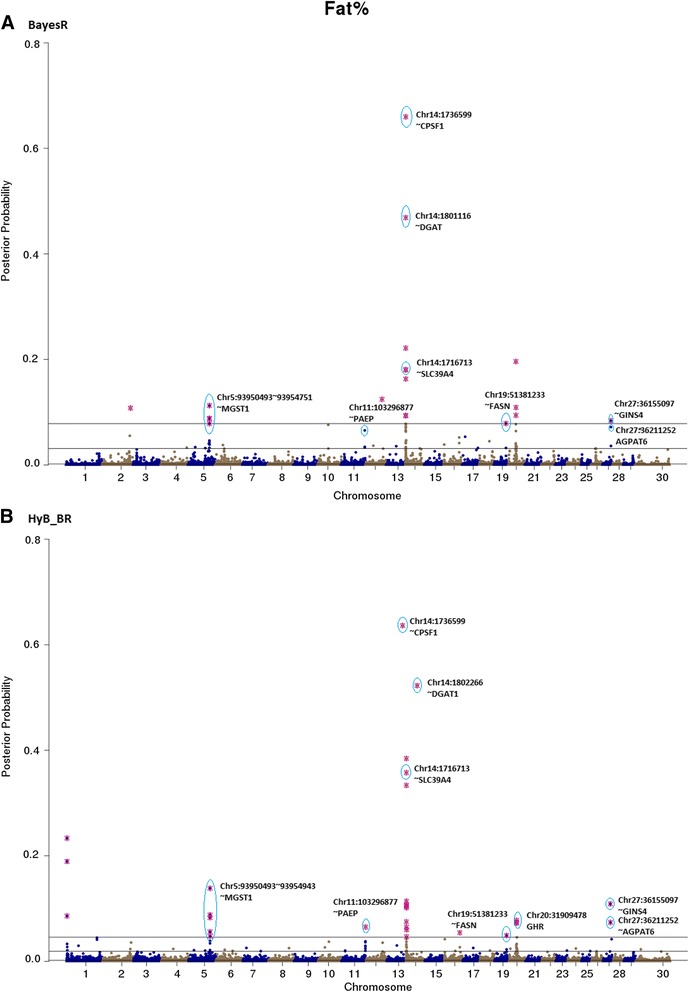
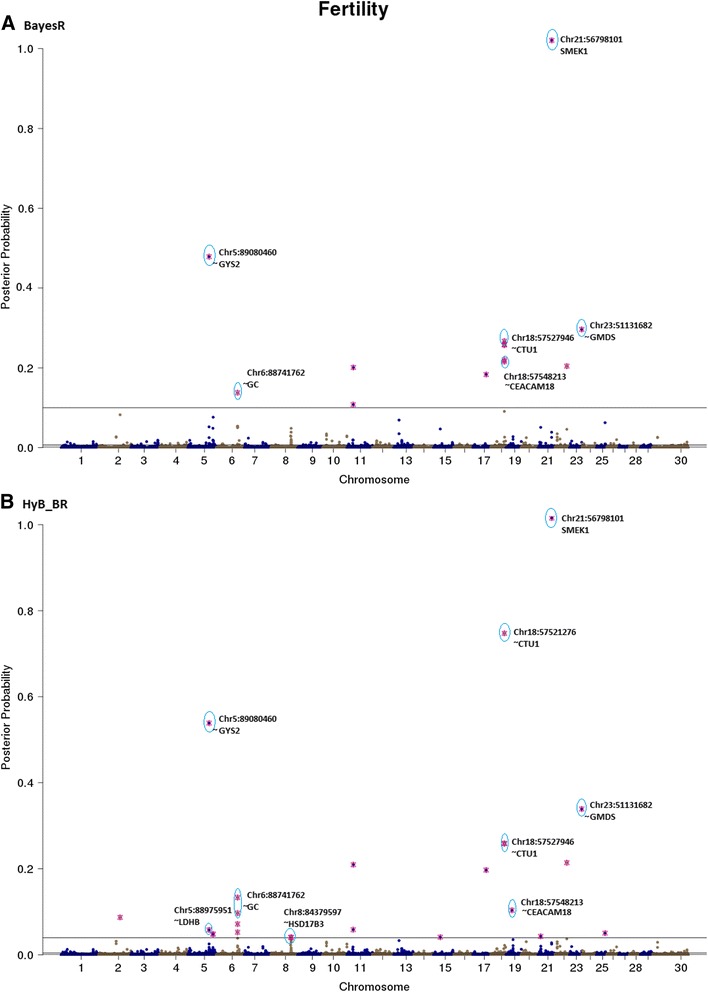
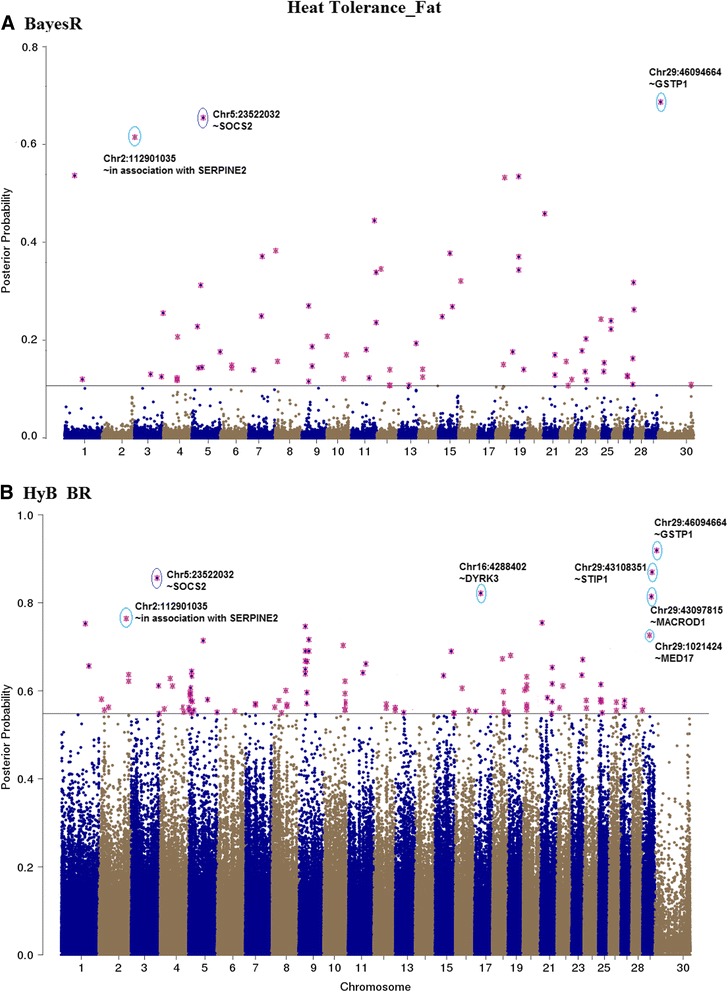
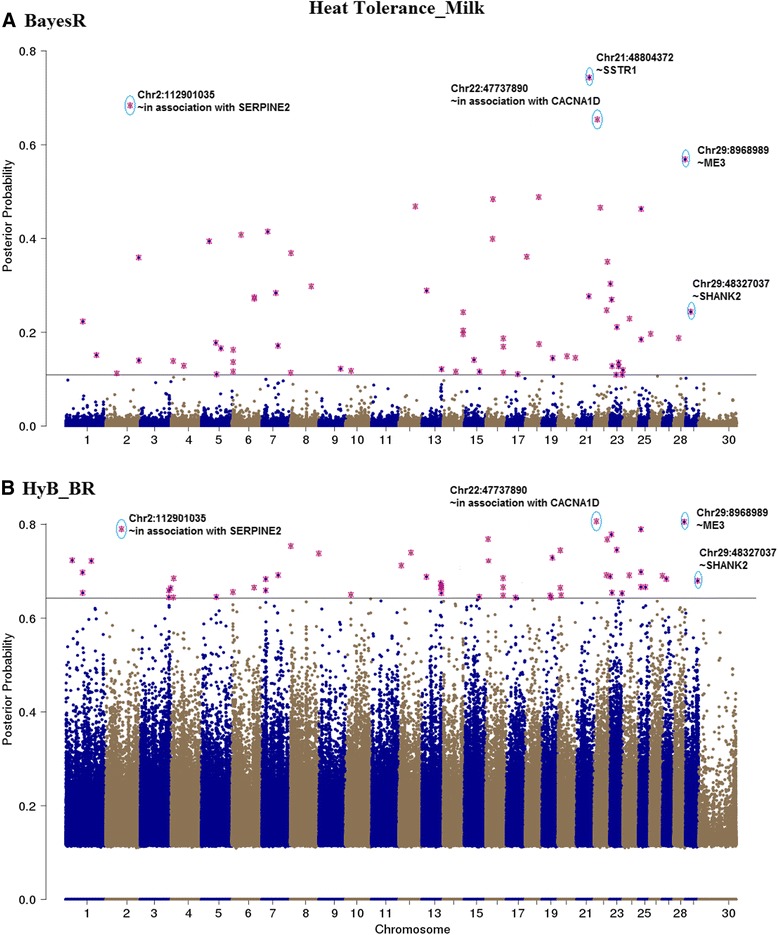
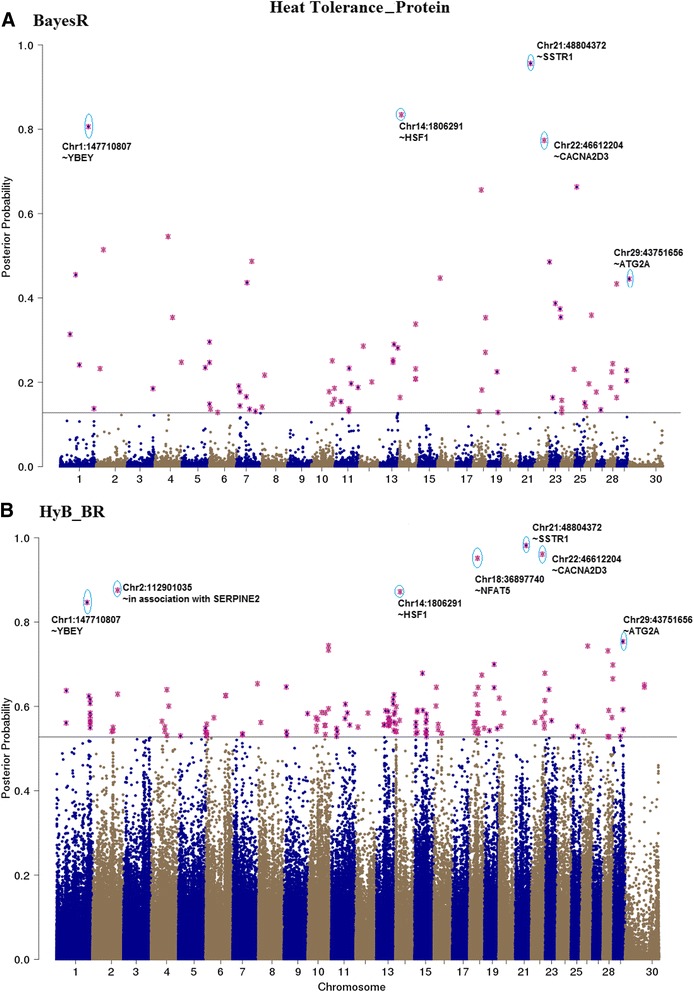
References
-
- IM ML, Hayes BJ, CJ VJ, Kemper KE, Haile-Mariam M, Bowman PJ, Schrooten C, Goddard ME. A Bayesian analysis to exploit imputed sequence variants for QTL discovery. In: Proceedings on 10th World Congress of Genetics Applied to Livestock Production: 2014. Vancouver, BC, Canada; 2014. p. 193.
-
- MacLeod IM, Bowman PJ, Vander Jagt CJ, Haile-Mariam M, Kemper KE, Chamberlain AJ, Schrooten C, Hayes BJ, Goddard ME. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genomics. 2016;17(1):1–21. doi: 10.1186/s12864-016-2443-6. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
