

Bacteriophages of Gordonia spp. Display a Spectrum of Diversity and Genetic Relationships
- PMID: 28811342
- PMCID: PMC5559632
- DOI: 10.1128/mBio.01069-17
Bacteriophages of Gordonia spp. Display a Spectrum of Diversity and Genetic Relationships
Abstract
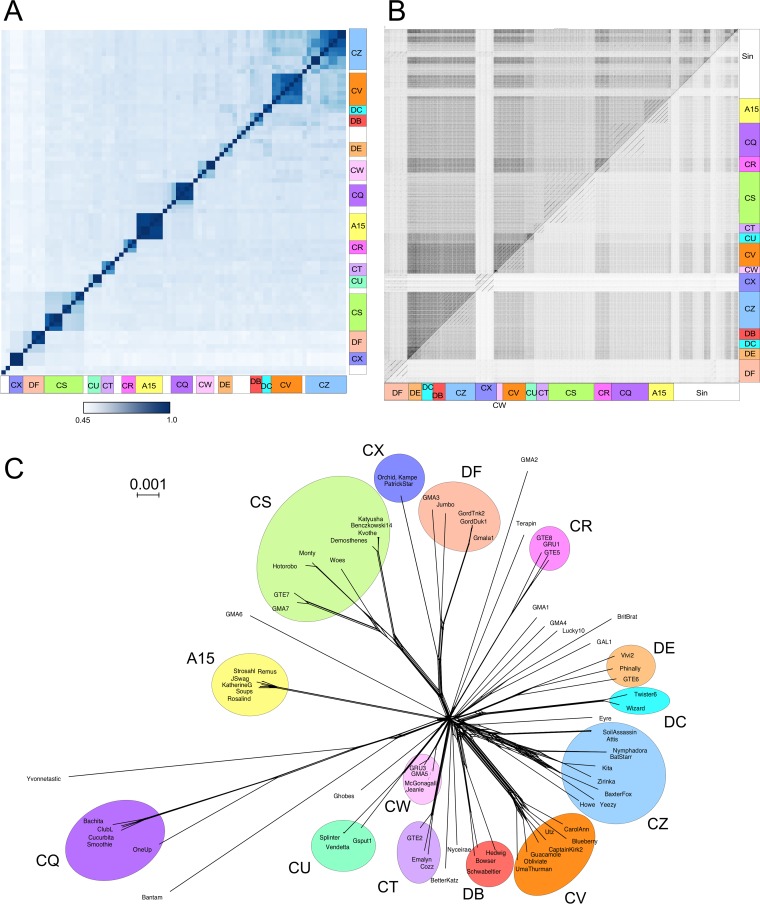
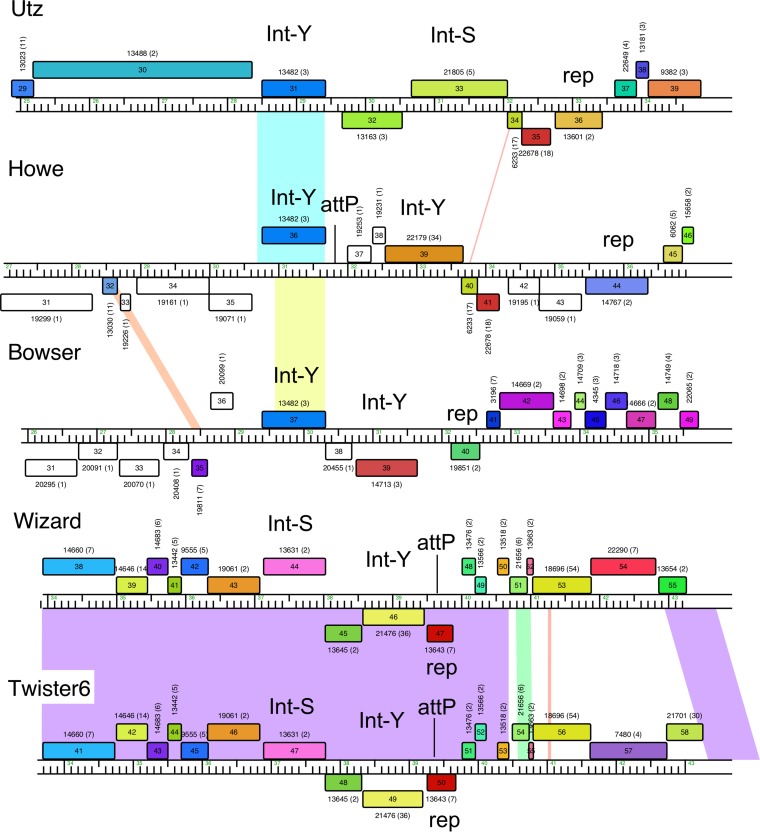
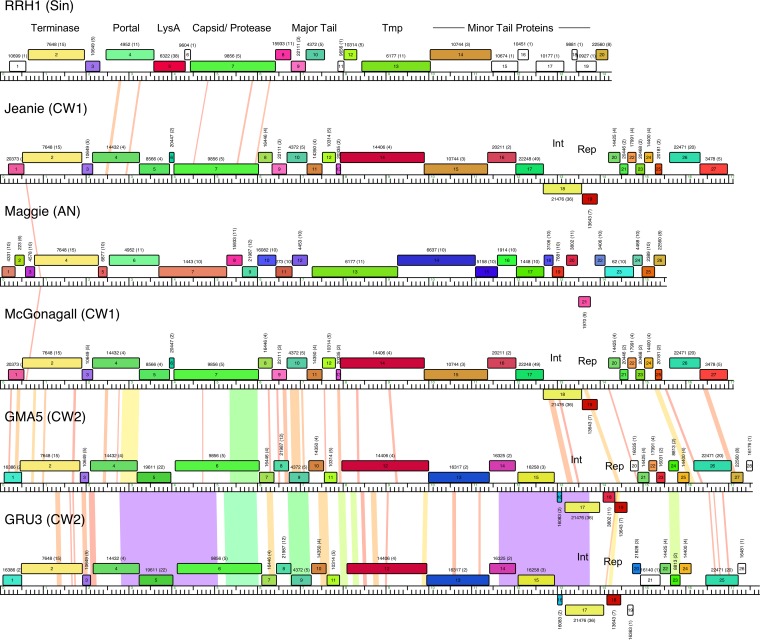
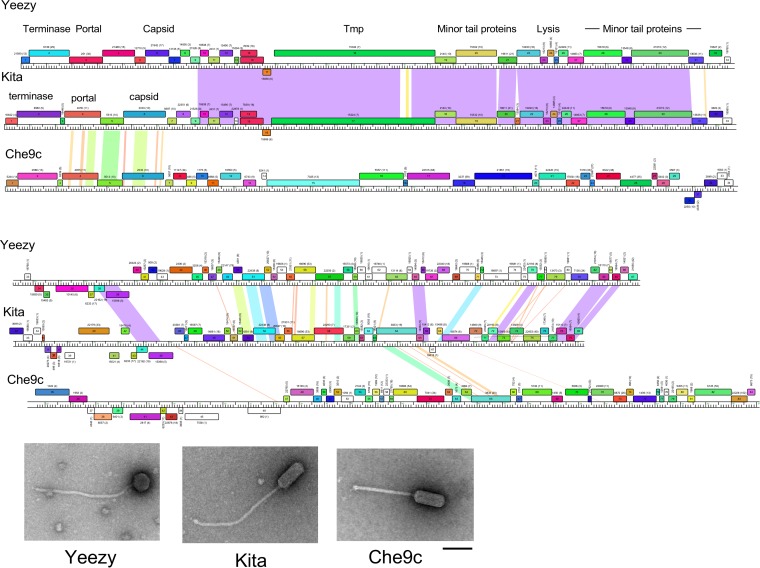
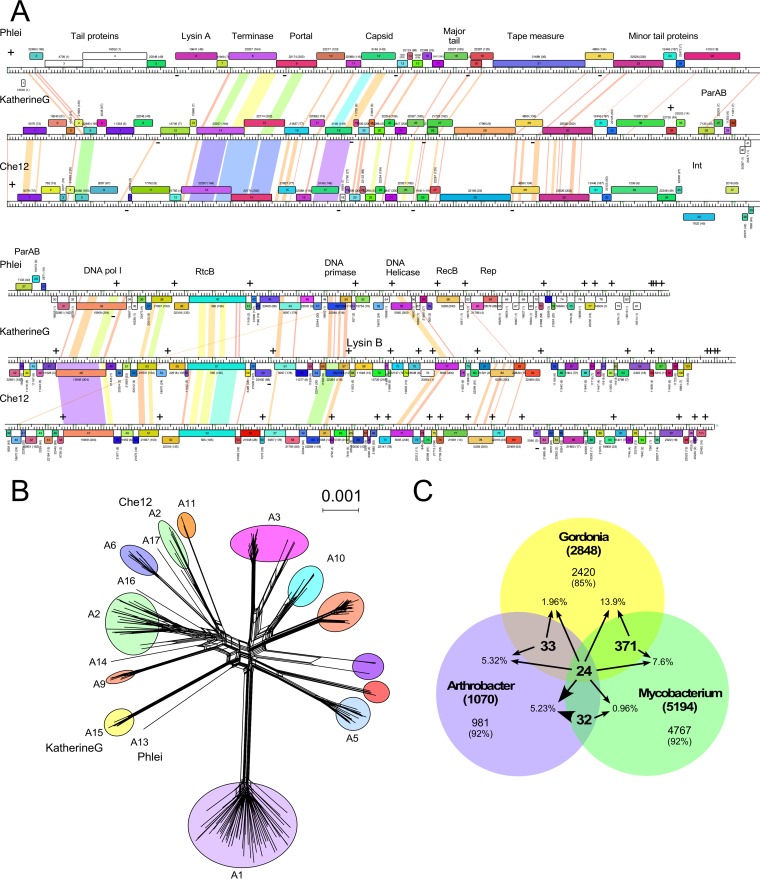
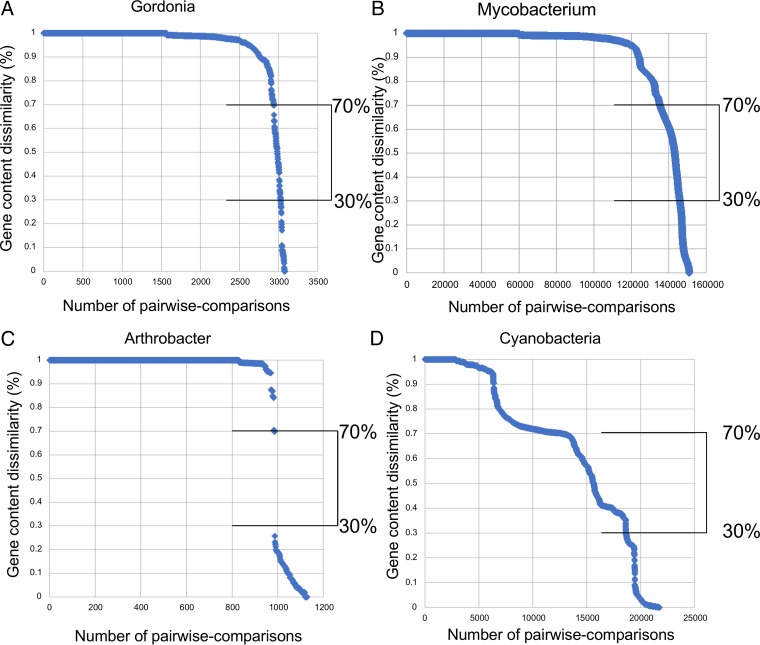
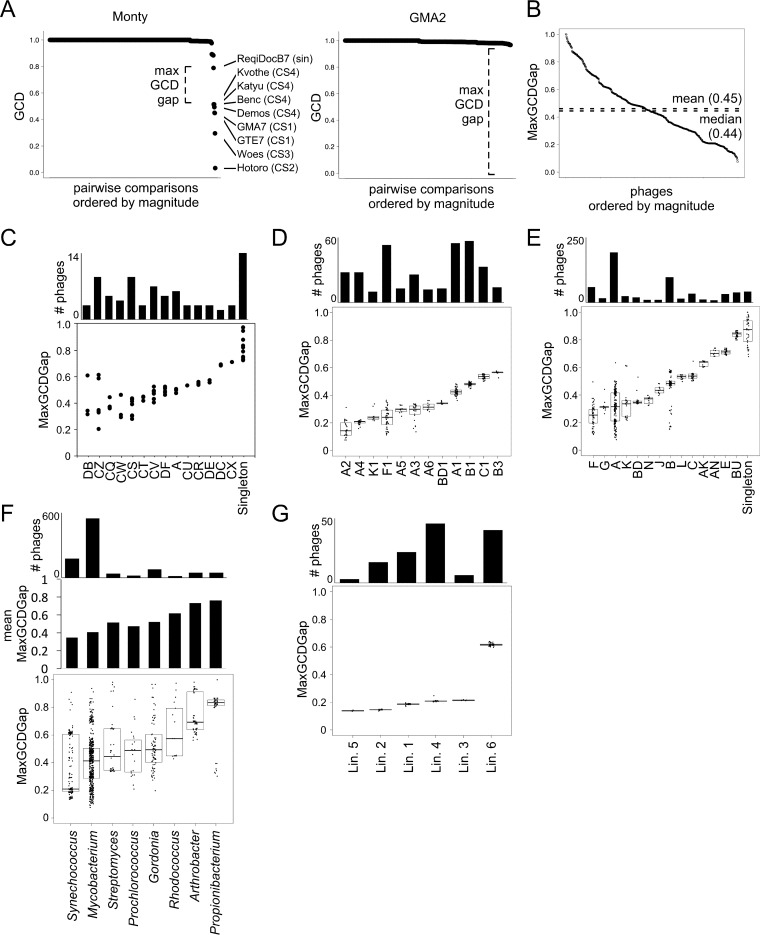
The global bacteriophage population is large, dynamic, old, and highly diverse genetically. Many phages are tailed and contain double-stranded DNA, but these remain poorly characterized genomically. A collection of over 1,000 phages infecting Mycobacterium smegmatis reveals the diversity of phages of a common bacterial host, but their relationships to phages of phylogenetically proximal hosts are not known. Comparative sequence analysis of 79 phages isolated on Gordonia shows these also to be diverse and that the phages can be grouped into 14 clusters of related genomes, with an additional 14 phages that are "singletons" with no closely related genomes. One group of six phages is closely related to Cluster A mycobacteriophages, but the other Gordonia phages are distant relatives and share only 10% of their genes with the mycobacteriophages. The Gordonia phage genomes vary in genome length (17.1 to 103.4 kb), percentage of GC content (47 to 68.8%), and genome architecture and contain a variety of features not seen in other phage genomes. Like the mycobacteriophages, the highly mosaic Gordonia phages demonstrate a spectrum of genetic relationships. We show this is a general property of bacteriophages and suggest that any barriers to genetic exchange are soft and readily violable.IMPORTANCE Despite the numerical dominance of bacteriophages in the biosphere, there is a dearth of complete genomic sequences. Current genomic information reveals that phages are highly diverse genomically and have mosaic architectures formed by extensive horizontal genetic exchange. Comparative analysis of 79 phages of Gordonia shows them to not only be highly diverse, but to present a spectrum of relatedness. Most are distantly related to phages of the phylogenetically proximal host Mycobacterium smegmatis, although one group of Gordonia phages is more closely related to mycobacteriophages than to the other Gordonia phages. Phage genome sequence space remains largely unexplored, but further isolation and genomic comparison of phages targeted at related groups of hosts promise to reveal pathways of bacteriophage evolution.
Keywords: Gordonia; bacteriophage genetics; bacteriophages.
Copyright © 2017 Pope et al.
Figures
References
-
- Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, Brucker W, Kumar V, Kandasamy J, Keenan L, Bardarov S, Kriakov J, Lawrence JG, Jacobs WR Jr, Hendrix RW, Hatfull GF. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171–182. doi: 10.1016/S0092-8674(03)00233-2. - DOI - PubMed
-
- Pope WH, Bowman CA, Russell DA, Jacobs-Sera D, Asai DJ, Cresawn SG, Jacobs WR, Hendrix RW, Lawrence JG, Hatfull GF, Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science, Phage Hunters Integrating Research and Education, Mycobacterial Genetics Course . 2015. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. eLife 4:e06416. doi: 10.7554/eLife.06416. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
