

The essential kinase ATR: ensuring faithful duplication of a challenging genome
- PMID: 28811666
- PMCID: PMC5796526
- DOI: 10.1038/nrm.2017.67
The essential kinase ATR: ensuring faithful duplication of a challenging genome
Erratum in
-
Publisher correction: The essential kinase ATR: ensuring faithful duplication of a challenging genome.Nat Rev Mol Cell Biol. 2017 Dec;18(12):783. doi: 10.1038/nrm.2017.116. Epub 2017 Nov 8. Nat Rev Mol Cell Biol. 2017. PMID: 29115300 No abstract available.
Abstract
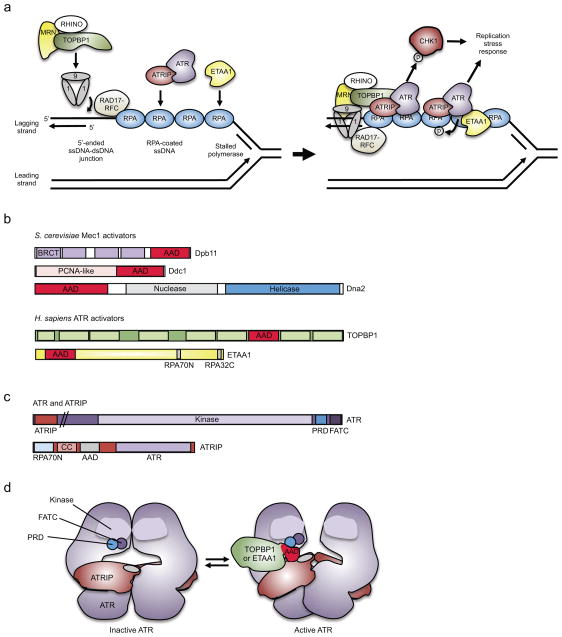
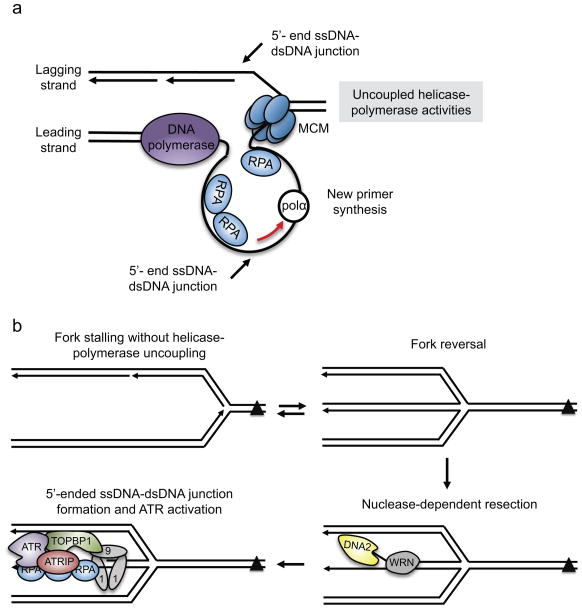
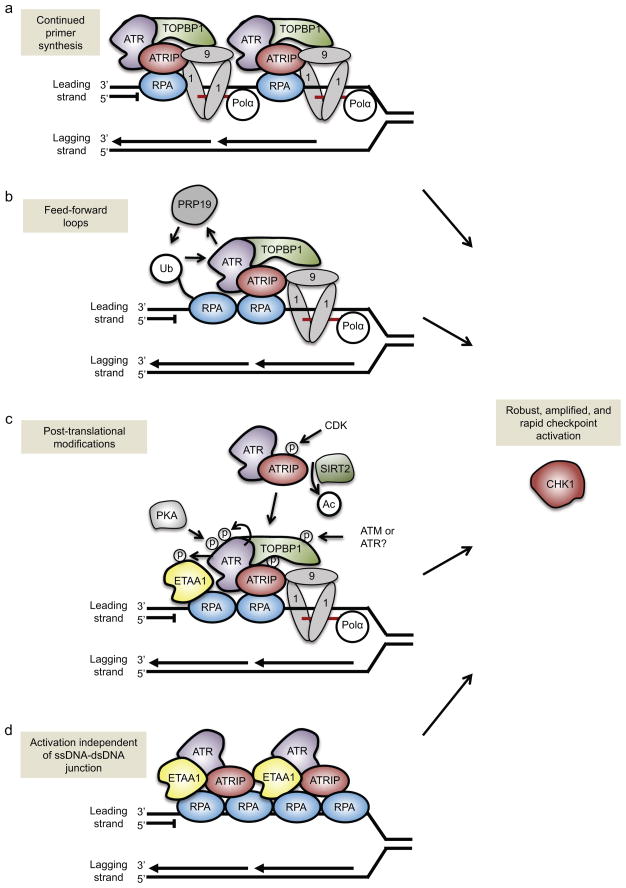
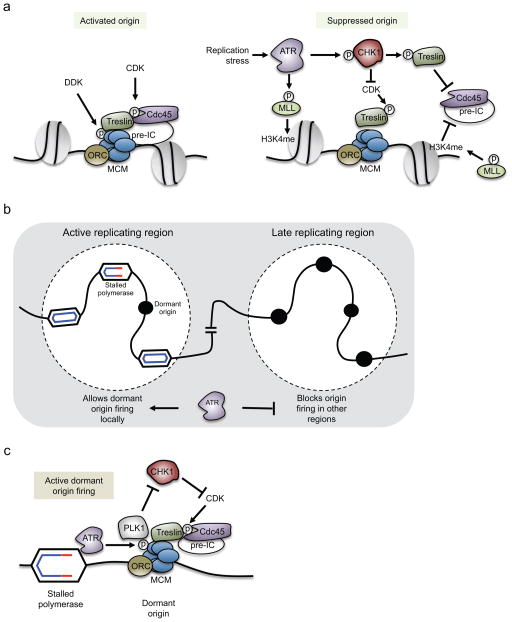
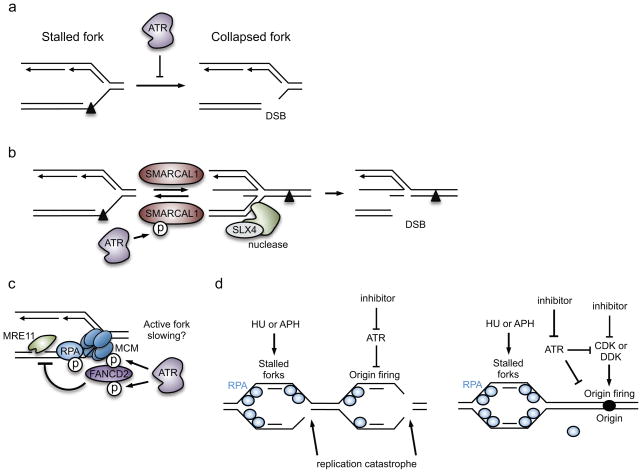
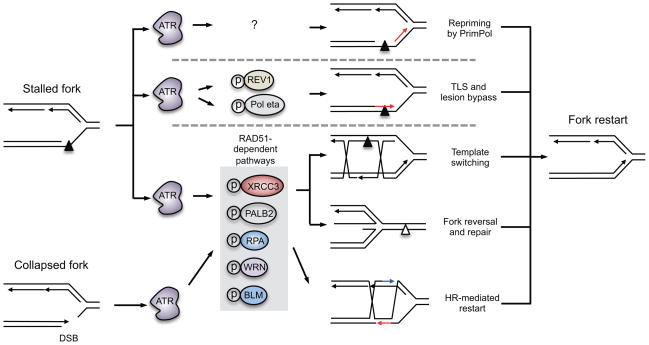
One way to preserve a rare book is to lock it away from all potential sources of damage. Of course, an inaccessible book is also of little use, and the paper and ink will continue to degrade with age in any case. Like a book, the information stored in our DNA needs to be read, but it is also subject to continuous assault and therefore needs to be protected. In this Review, we examine how the replication stress response that is controlled by the kinase ataxia telangiectasia and Rad3-related (ATR) senses and resolves threats to DNA integrity so that the DNA remains available to read in all of our cells. We discuss the multiple data that have revealed an elegant yet increasingly complex mechanism of ATR activation. This involves a core set of components that recruit ATR to stressed replication forks, stimulate kinase activity and amplify ATR signalling. We focus on the activities of ATR in the control of cell cycle checkpoints, origin firing and replication fork stability, and on how proper regulation of these processes is crucial to ensure faithful duplication of a challenging genome.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- de Klein A, et al. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479–482. Demonstrated that ATR is essential for viability in vivo. - PubMed
-
- O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nature Genet. 2003;33:497–501. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
