

Selected missense mutations impair frataxin processing in Friedreich ataxia
- PMID: 28812047
- PMCID: PMC5553228
- DOI: 10.1002/acn3.433
Selected missense mutations impair frataxin processing in Friedreich ataxia
Erratum in
-
Erratum: Selected missense mutations impair frataxin processing in Friedreich ataxia.Ann Clin Transl Neurol. 2017 Oct 10;4(10):764. doi: 10.1002/acn3.490. eCollection 2017 Oct. Ann Clin Transl Neurol. 2017. PMID: 29046887 Free PMC article.
Abstract
Objective: Frataxin (FXN) is a highly conserved mitochondrial protein. Reduced FXN levels cause Friedreich ataxia, a recessive neurodegenerative disease. Typical patients carry GAA repeat expansions on both alleles, while a subgroup of patients carry a missense mutation on one allele and a GAA repeat expansion on the other. Here, we report that selected disease-related FXN missense mutations impair FXN localization, interaction with mitochondria processing peptidase, and processing.
Methods: Immunocytochemical studies and subcellular fractionation were performed to study FXN import into the mitochondria and examine the mechanism by which mutations impair FXN processing. Coimmunoprecipitation was performed to study the interaction between FXN and mitochondrial processing peptidase. A proteasome inhibitor was used to model traditional therapeutic strategies. In addition, clinical profiles of subjects with and without point mutations were compared in a large natural history study.
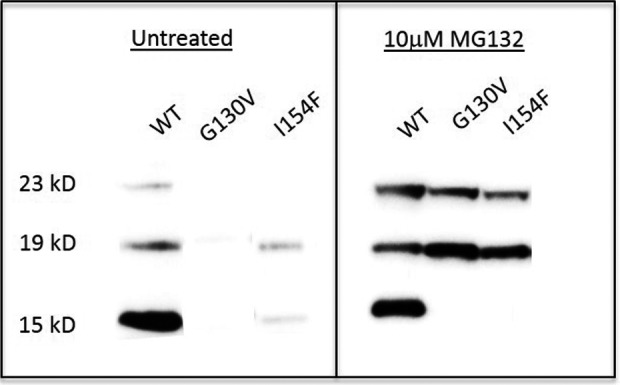
Results: FXNI154F and FXNG130V missense mutations decrease FXN 81-210 levels compared with FXNWT, FXNR165C, and FXNW155R, but do not block its association with mitochondria. FXNI154F and FXNG130V also impair FXN maturation and enhance the binding between FXN 42-210 and mitochondria processing peptidase. Furthermore, blocking proteosomal degradation does not increase FXN 81-210 levels. Additionally, impaired FXN processing also occurs in fibroblasts from patients with FXNG130V. Finally, clinical data from patients with FXNG130V and FXNI154F mutations demonstrates a lower severity compared with other individuals with Friedreich ataxia.
Interpretation: These data suggest that the effects on processing associated with FXNG130V and FXNI154F mutations lead to higher levels of partially processed FXN, which may contribute to the milder clinical phenotypes in these patients.
Figures





References
-
- Bidichandani S, Delatycki M. Friedreich Ataxia. Gene Reviews 2014.
-
- Marmolino D. Friederich's ataxia: past, present, and future. Brains Res Rev J 2011;2:311–330. - PubMed
-
- Campuzano V, Montermini L, Dolores Molto M, et al. Friedreich's ataxia: autosomal recessive disease caused by an lntronic GAA triplet repeat expansion. Science 1996;271:1423–1427. - PubMed
-
- Gonzalez‐Cabo P, Vazquez‐Manrique V, Adelaida Garcia‐Gimeno M, et al. Frataxin interacts functionally with mitochondrial electron transport chain proteins. Hum Mol Genet 2005;14:2091–2098. - PubMed
-
- Dürr A, Cossee M, Agid Y, et al. Clinical and genetic abnormalities in patients with Friedreich's ataxia. N Engl J Med 1996;335:1169–1175. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
