

Antibiotics Disrupt Coordination between Transcriptional and Phenotypic Stress Responses in Pathogenic Bacteria
- PMID: 28813680
- PMCID: PMC5584877
- DOI: 10.1016/j.celrep.2017.07.062
Antibiotics Disrupt Coordination between Transcriptional and Phenotypic Stress Responses in Pathogenic Bacteria
Abstract
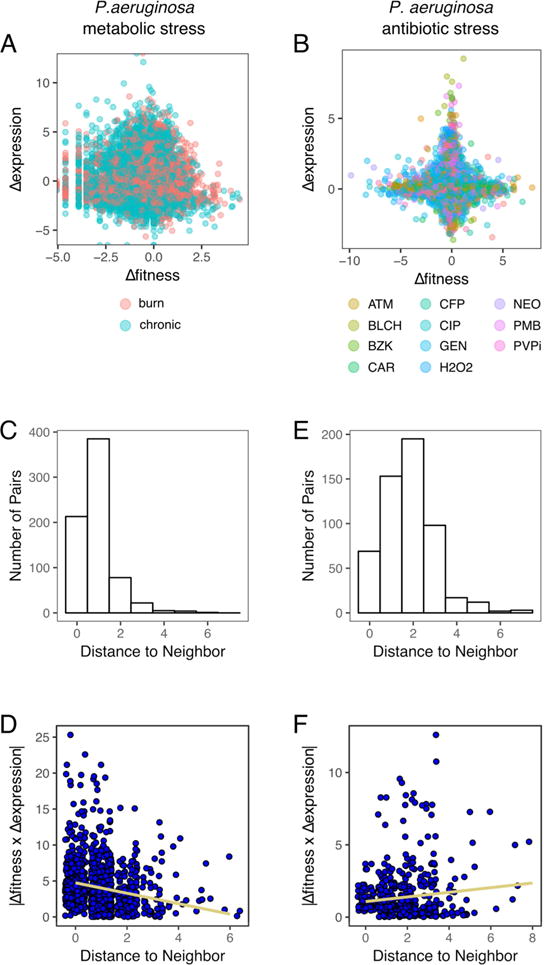
Bacterial genes that change in expression upon environmental disturbance have commonly been seen as those that must also phenotypically matter. However, several studies suggest that differentially expressed genes are rarely phenotypically important. We demonstrate, for Gram-positive and Gram-negative bacteria, that these seemingly uncoordinated gene sets are involved in responses that can be linked through topological network analysis. However, the level of coordination is stress dependent. While a well-coordinated response is triggered in response to nutrient stress, antibiotics trigger an uncoordinated response in which transcriptionally and phenotypically important genes are neither linked spatially nor in their magnitude. Moreover, a gene expression meta-analysis reveals that genes with large fitness changes during stress have low transcriptional variation across hundreds of other conditions, and vice versa. Our work suggests that cellular responses can be understood through network models that incorporate regulatory and genetic relationships, which could aid drug target predictions and genetic network engineering.
Keywords: Pseudomonas; RNA-seq; Streptococcus; Tn-seq; data integration; metabolic modeling; stress response; systems biology.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures






References
-
- Aaberge IS, Eng J, Lermark G, Løvik M. Virulence of Streptococcus pneumoniae in mice: a standardized method for preparation and frozen storage of the experimental bacterial inoculum. Microbial Pathogenesis. 1995;18:141–152. - PubMed
-
- Bao H, Kommadath A, Liang G, Sun X, Arantes AS, Tuggle CK, Bearson SMD, Plastow GS, Stothard P, Guan LL. Genome-wide whole blood microRNAome and transcriptome analyses reveal miRNA-mRNA regulated host response to foodborne pathogen Salmonella infection in swine. Sci Rep. 2015;5:e12620. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
