

Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations
- PMID: 28814792
- PMCID: PMC5559603
- DOI: 10.1038/s41467-017-00366-1
Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations
Abstract
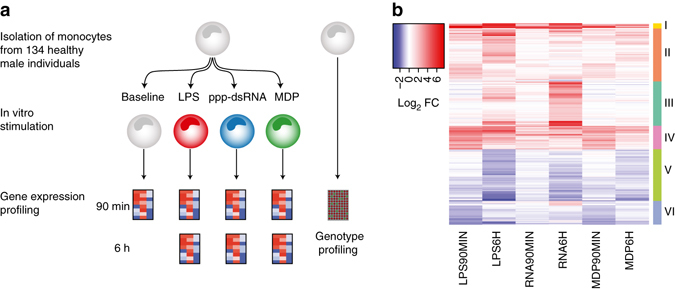
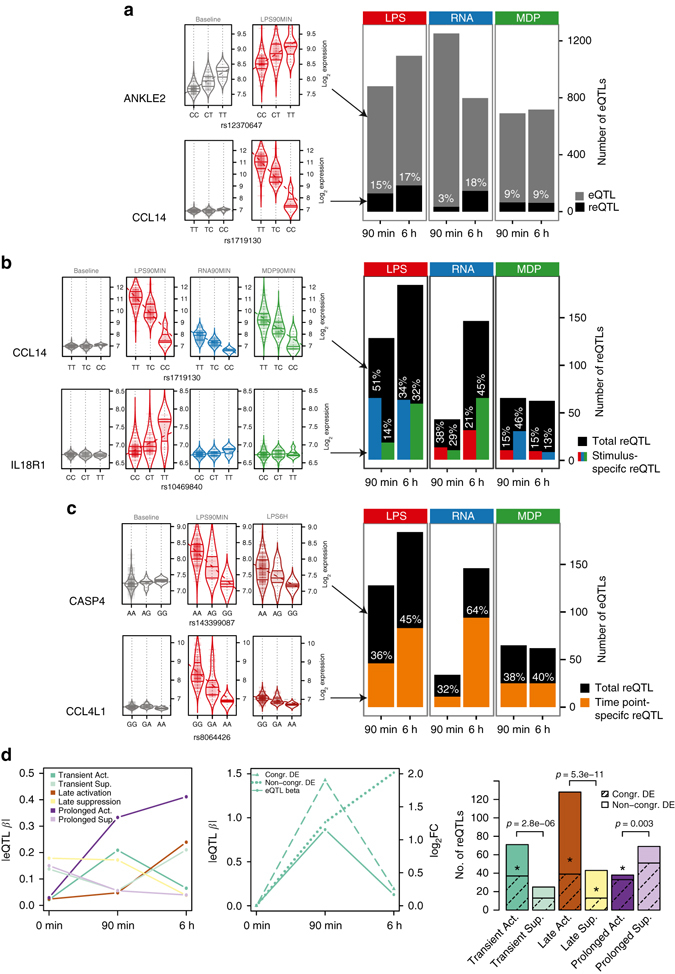
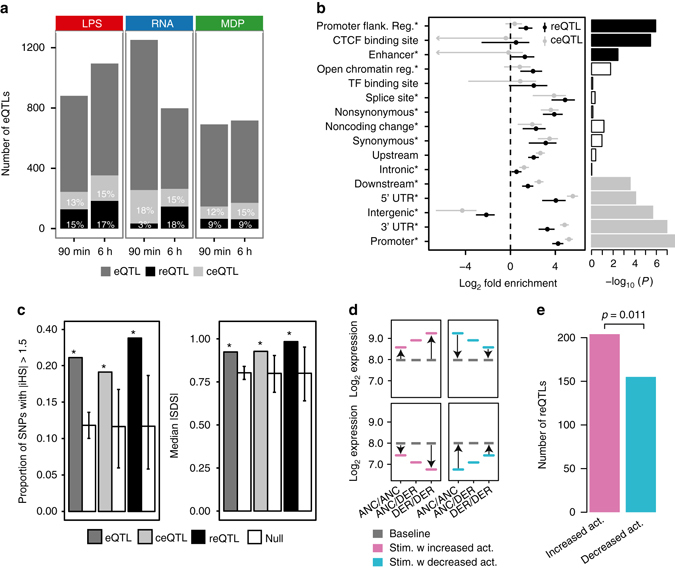
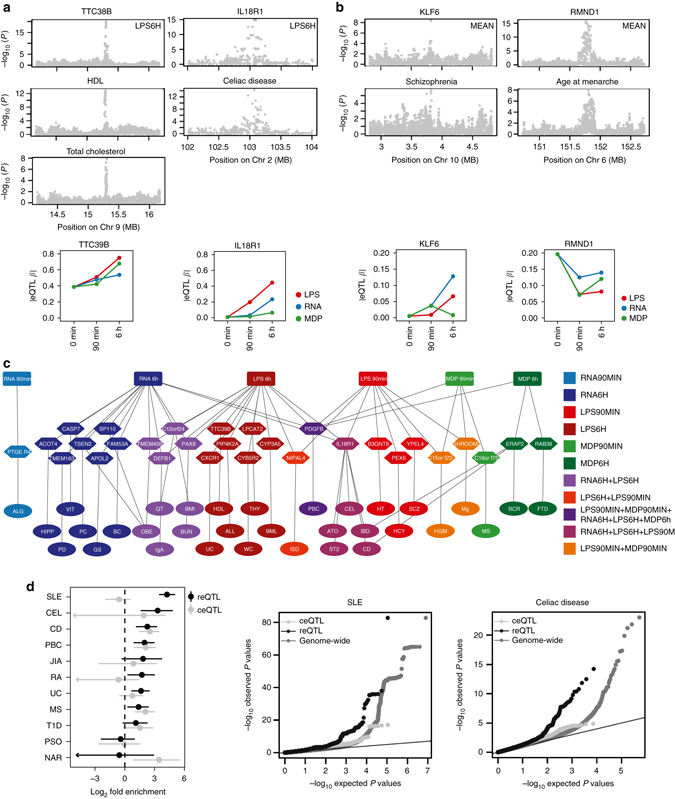
The immune system plays a major role in human health and disease, and understanding genetic causes of interindividual variability of immune responses is vital. Here, we isolate monocytes from 134 genotyped individuals, stimulate these cells with three defined microbe-associated molecular patterns (LPS, MDP, and 5'-ppp-dsRNA), and profile the transcriptomes at three time points. Mapping expression quantitative trait loci (eQTL), we identify 417 response eQTLs (reQTLs) with varying effects between conditions. We characterize the dynamics of genetic regulation on early and late immune response and observe an enrichment of reQTLs in distal cis-regulatory elements. In addition, reQTLs are enriched for recent positive selection with an evolutionary trend towards enhanced immune response. Finally, we uncover reQTL effects in multiple GWAS loci and show a stronger enrichment for response than constant eQTLs in GWAS signals of several autoimmune diseases. This demonstrates the importance of infectious stimuli in modifying genetic predisposition to disease.Insight into the genetic influence on the immune response is important for the understanding of interindividual variability in human pathologies. Here, the authors generate transcriptome data from human blood monocytes stimulated with various immune stimuli and provide a time-resolved response eQTL map.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
