

Monoallelic and biallelic CREB3L1 variant causes mild and severe osteogenesis imperfecta, respectively
- PMID: 28817112
- PMCID: PMC5816725
- DOI: 10.1038/gim.2017.115
Monoallelic and biallelic CREB3L1 variant causes mild and severe osteogenesis imperfecta, respectively
Abstract
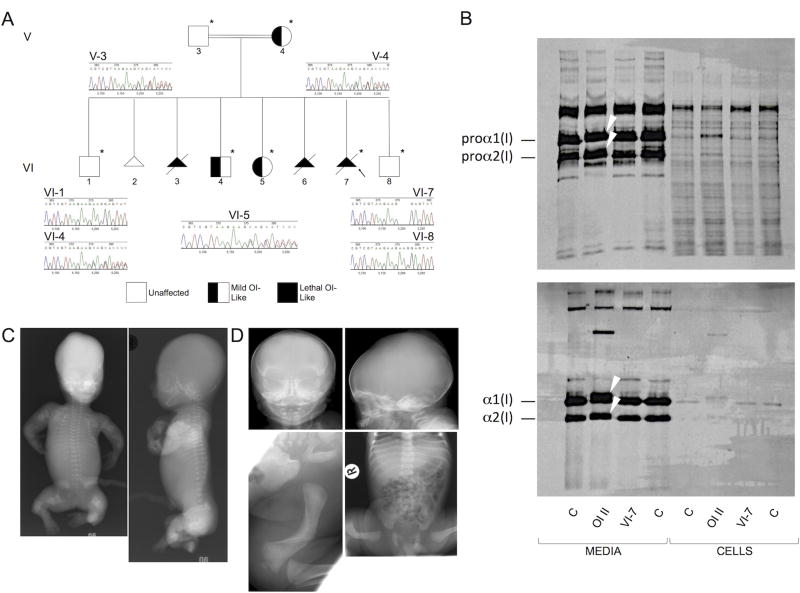
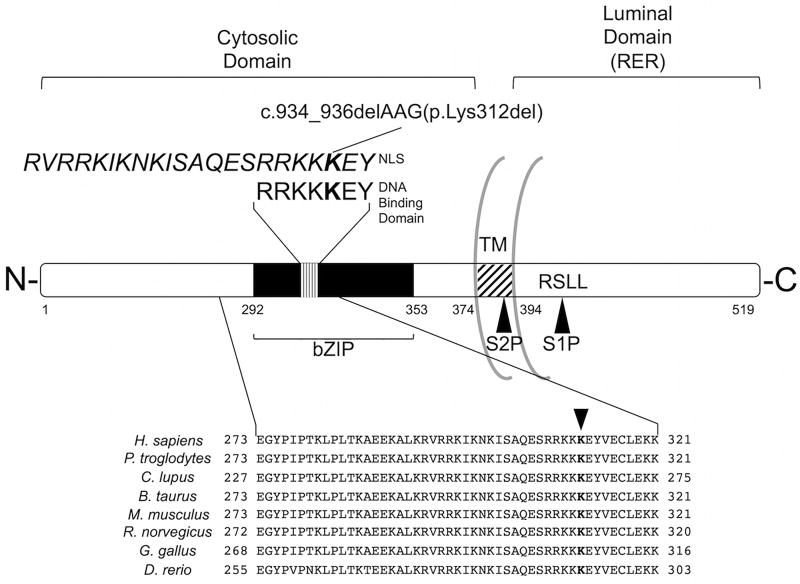
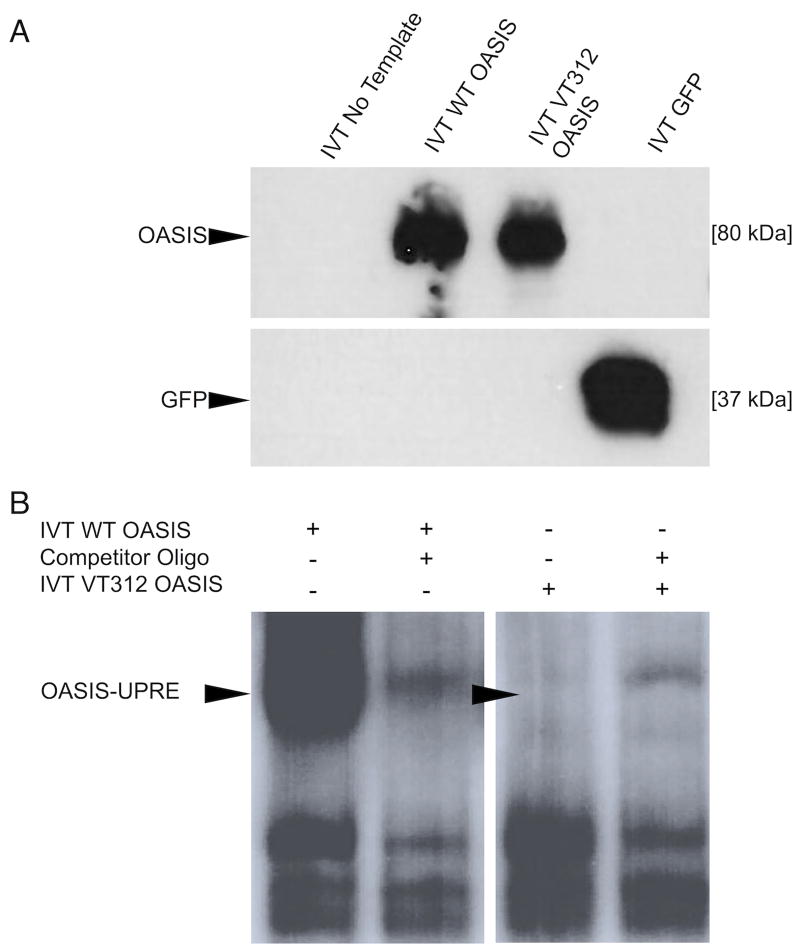
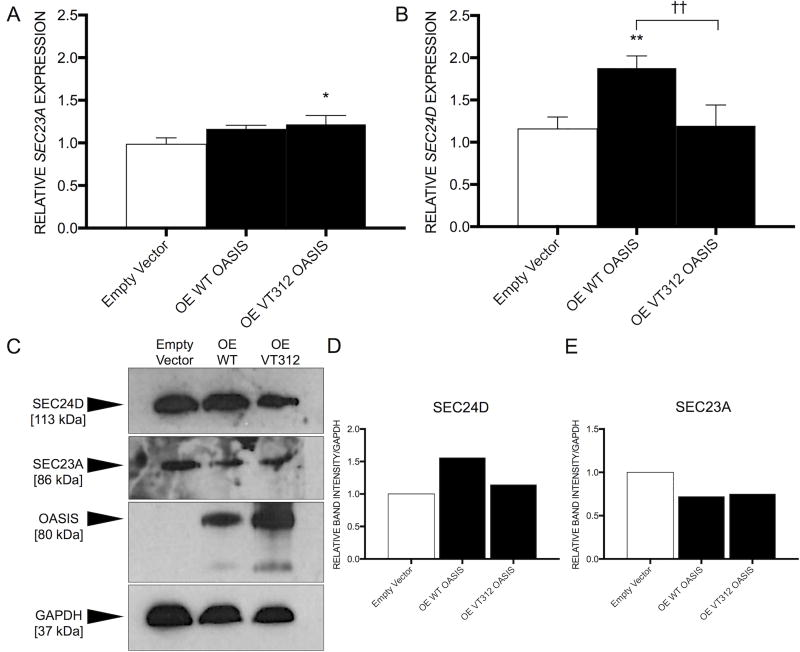
PurposeOsteogenesis imperfecta (OI) is a heritable skeletal dysplasia. Dominant pathogenic variants in COL1A1 and COL1A2 explain the majority of OI cases. At least 15 additional genes have been identified, but those still do not account for all OI phenotypes that present. We sought the genetic cause of mild and lethal OI phenotypes in an unsolved family.MethodsWe performed exome sequencing on seven members of the family, both affected and unaffected.ResultsWe identified a variant in cyclic AMP responsive element binding protein 3-like 1 (CREB3L1) in a consanguineous family. The variant caused a prenatal/perinatal lethal OI in homozygotes, similar to that seen in OI type II as a result of mutations in type I collagen genes, and a mild phenotype (fractures, blue sclerae) in multiple heterozygous family members. CREB3L1 encodes old astrocyte specifically induced substance (OASIS), an endoplasmic reticulum stress transducer. The variant disrupts a DNA-binding site and prevents OASIS from acting on its transcriptional targets including SEC24D, which encodes a component of the coat protein II complex.ConclusionThis report confirms that CREB3L1 is an OI-related gene and suggests the pathogenic mechanism of CREB3L1-associated OI involves the altered regulation of proteins involved in cellular secretion.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
