

Pharmacological and Activated Fibroblast Targeting of Gβγ-GRK2 After Myocardial Ischemia Attenuates Heart Failure Progression
- PMID: 28818206
- PMCID: PMC5564231
- DOI: 10.1016/j.jacc.2017.06.049
Pharmacological and Activated Fibroblast Targeting of Gβγ-GRK2 After Myocardial Ischemia Attenuates Heart Failure Progression
Abstract
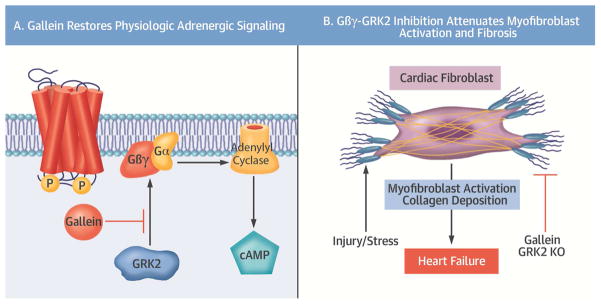
Background: Cardiac fibroblasts are a critical cell population responsible for myocardial extracellular matrix homeostasis. Upon injury or pathological stimulation, these cells transform to an activated myofibroblast state and play a fundamental role in myocardial fibrosis and remodeling. Chronic sympathetic overstimulation, a hallmark of heart failure (HF), induces pathological signaling through G protein βγ (Gβγ) subunits and their interaction with G protein-coupled receptor kinase 2 (GRK2).
Objectives: This study investigated the hypothesis that Gβγ-GRK2 inhibition and/or ablation after myocardial injury would attenuate pathological myofibroblast activation and cardiac remodeling.
Methods: The therapeutic potential of small molecule Gβγ-GRK2 inhibition, alone or in combination with activated fibroblast- or myocyte-specific GRK2 ablation-each initiated after myocardial ischemia-reperfusion (I/R) injury-was investigated to evaluate the possible salutary effects on post-I/R fibroblast activation, pathological remodeling, and cardiac dysfunction.
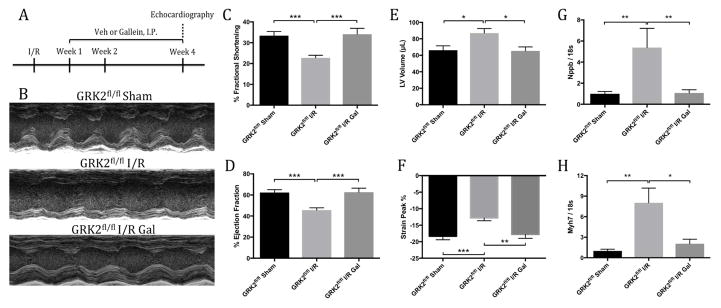
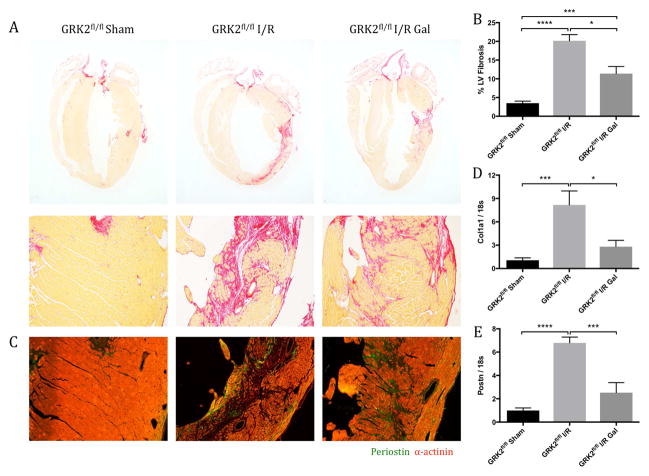
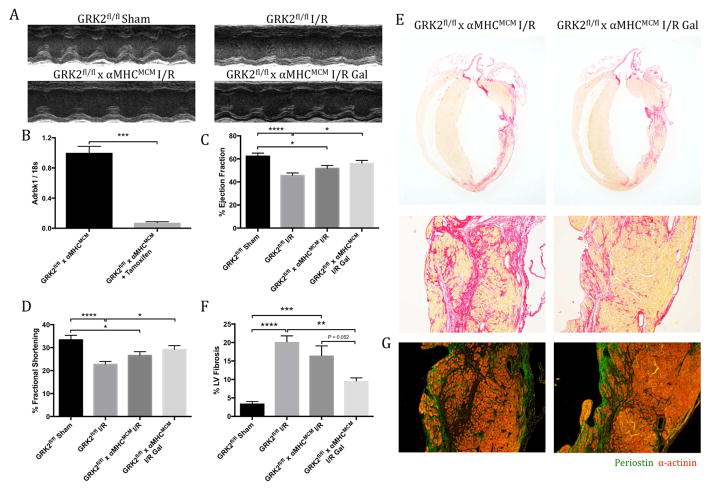
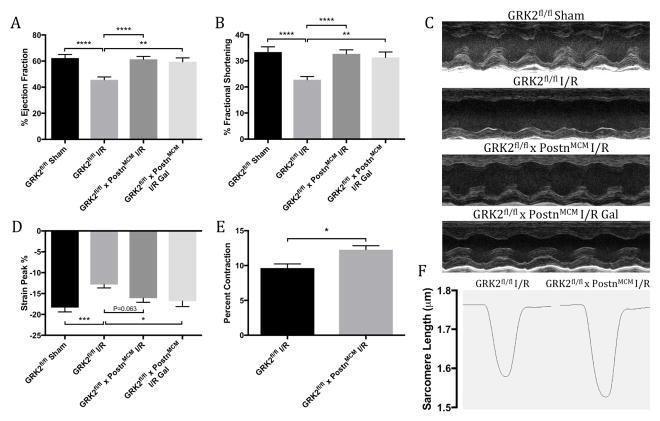
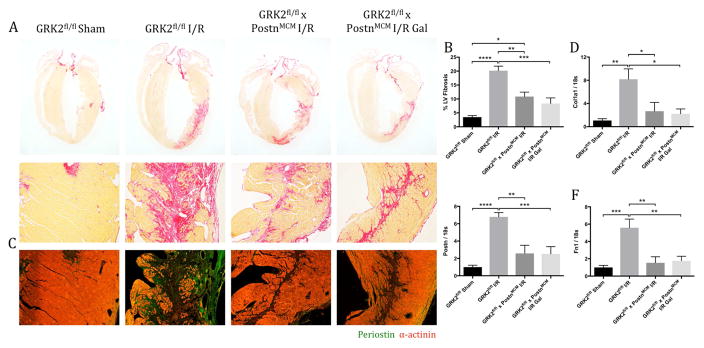
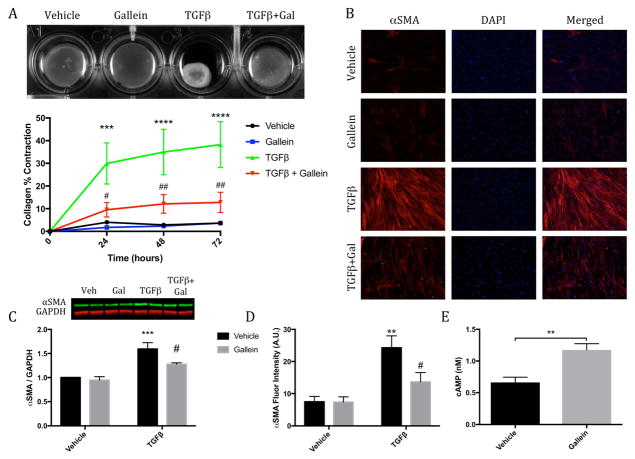
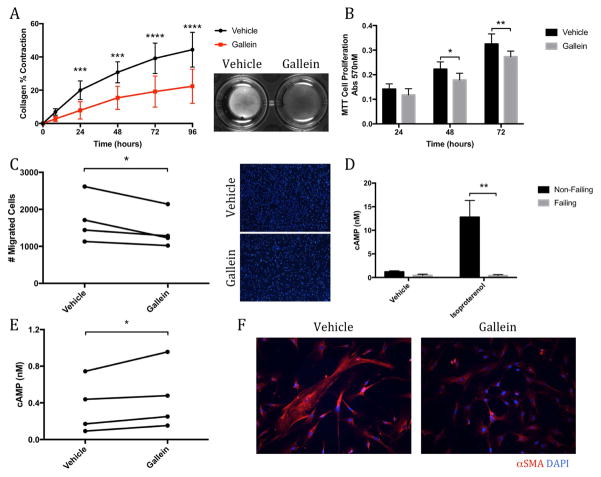
Results: Small molecule Gβγ-GRK2 inhibition initiated 1 week post-injury was cardioprotective in the I/R model of chronic HF, including preservation of cardiac contractility and a reduction in cardiac fibrotic remodeling. Systemic small molecule Gβγ-GRK2 inhibition initiated 1 week post-I/R in cardiomyocyte-restricted GRK2 ablated mice (also post-I/R) still demonstrated significant cardioprotection, which suggested a potential protective role beyond the cardiomyocyte. Inducible ablation of GRK2 in activated fibroblasts (i.e., myofibroblasts) post-I/R injury demonstrated significant functional cardioprotection with reduced myofibroblast transformation and fibrosis. Systemic small molecule Gβγ-GRK2 inhibition initiated 1 week post-I/R provided little to no further protection in mice with ablation of GRK2 in activated fibroblasts alone. Finally, Gβγ-GRK2 inhibition significantly attenuated activation characteristics of failing human cardiac fibroblasts isolated from end-stage HF patients.
Conclusions: These findings suggested consideration of a paradigm shift in the understanding of the therapeutic role of Gβγ-GRK2 inhibition in treating HF and the potential therapeutic role for Gβγ-GRK2 inhibition in limiting pathological myofibroblast activation, interstitial fibrosis, and HF progression.
Keywords: cardiac fibroblast; cardioprotection; fibrosis; remodeling.
Copyright © 2017 American College of Cardiology Foundation. Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
β-Adrenergic Blockade in Ischemia-Reperfusion Injury: βARKing Up a New Tree.J Am Coll Cardiol. 2017 Aug 22;70(8):972-974. doi: 10.1016/j.jacc.2017.06.062. J Am Coll Cardiol. 2017. PMID: 28818207 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
