

Comprehensive single-cell transcriptional profiling of a multicellular organism
- PMID: 28818938
- PMCID: PMC5894354
- DOI: 10.1126/science.aam8940
Comprehensive single-cell transcriptional profiling of a multicellular organism
Abstract
To resolve cellular heterogeneity, we developed a combinatorial indexing strategy to profile the transcriptomes of single cells or nuclei, termed sci-RNA-seq (single-cell combinatorial indexing RNA sequencing). We applied sci-RNA-seq to profile nearly 50,000 cells from the nematode Caenorhabditis elegans at the L2 larval stage, which provided >50-fold "shotgun" cellular coverage of its somatic cell composition. From these data, we defined consensus expression profiles for 27 cell types and recovered rare neuronal cell types corresponding to as few as one or two cells in the L2 worm. We integrated these profiles with whole-animal chromatin immunoprecipitation sequencing data to deconvolve the cell type-specific effects of transcription factors. The data generated by sci-RNA-seq constitute a powerful resource for nematode biology and foreshadow similar atlases for other organisms.
Copyright © 2017 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
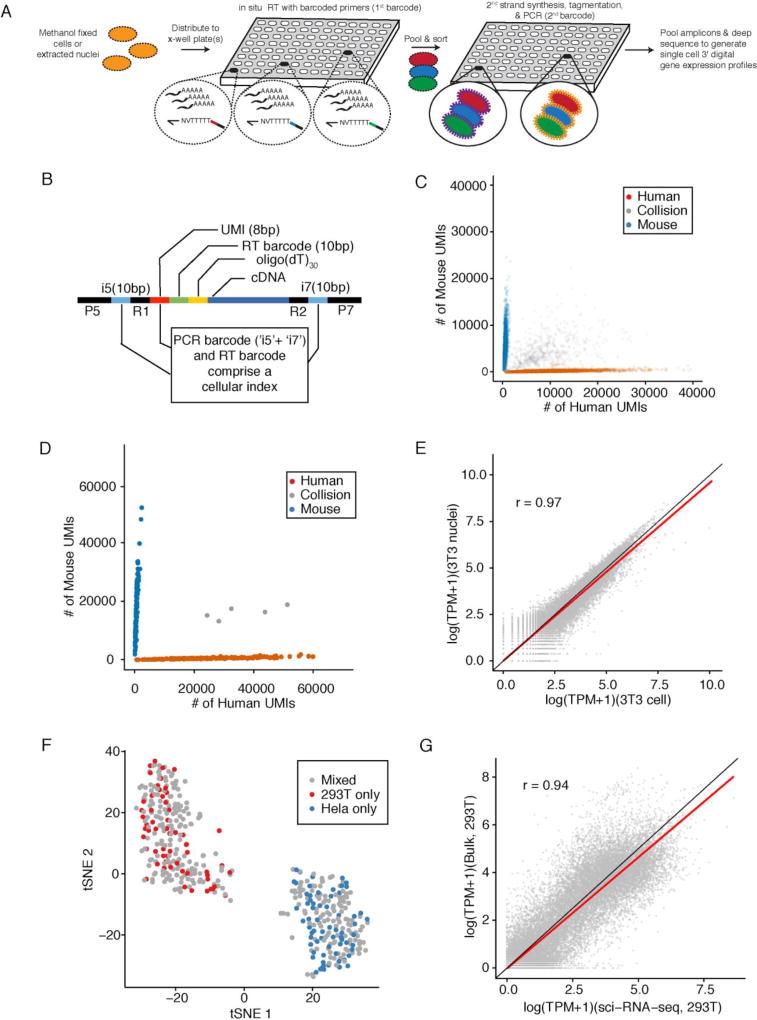
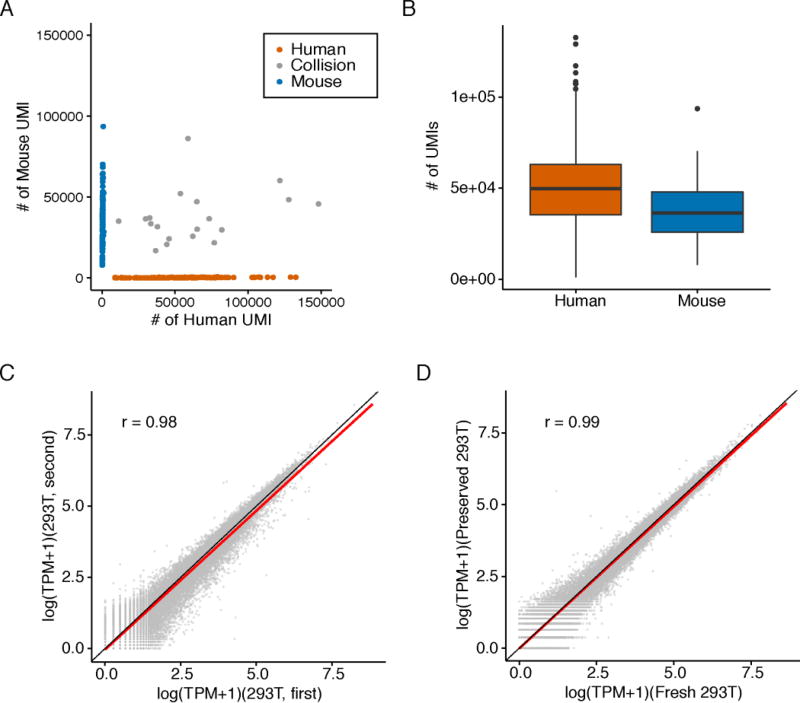
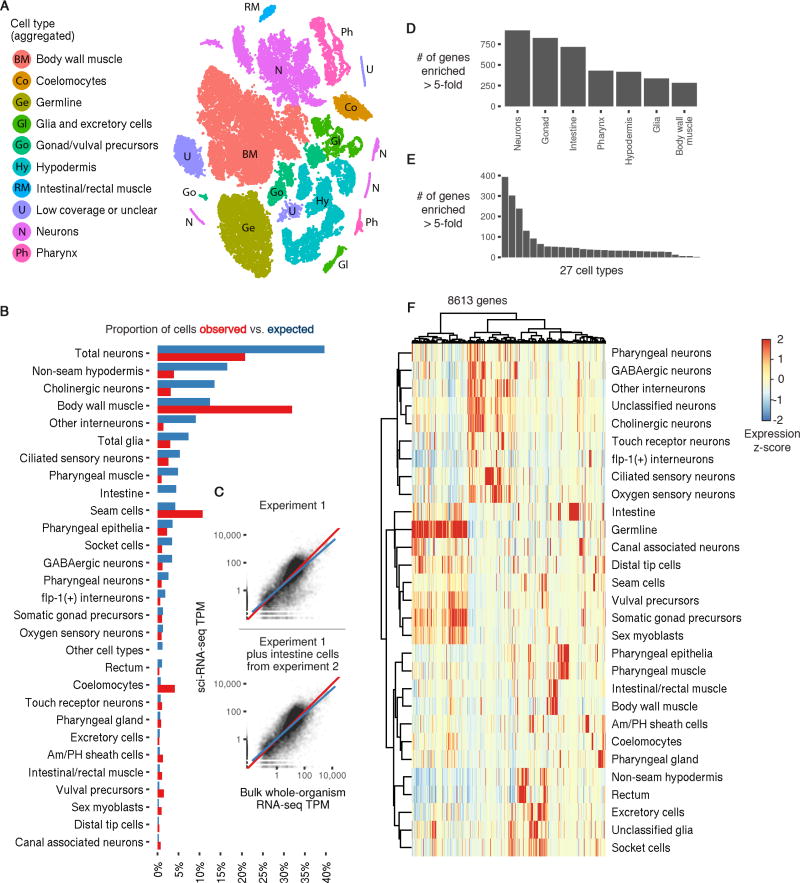
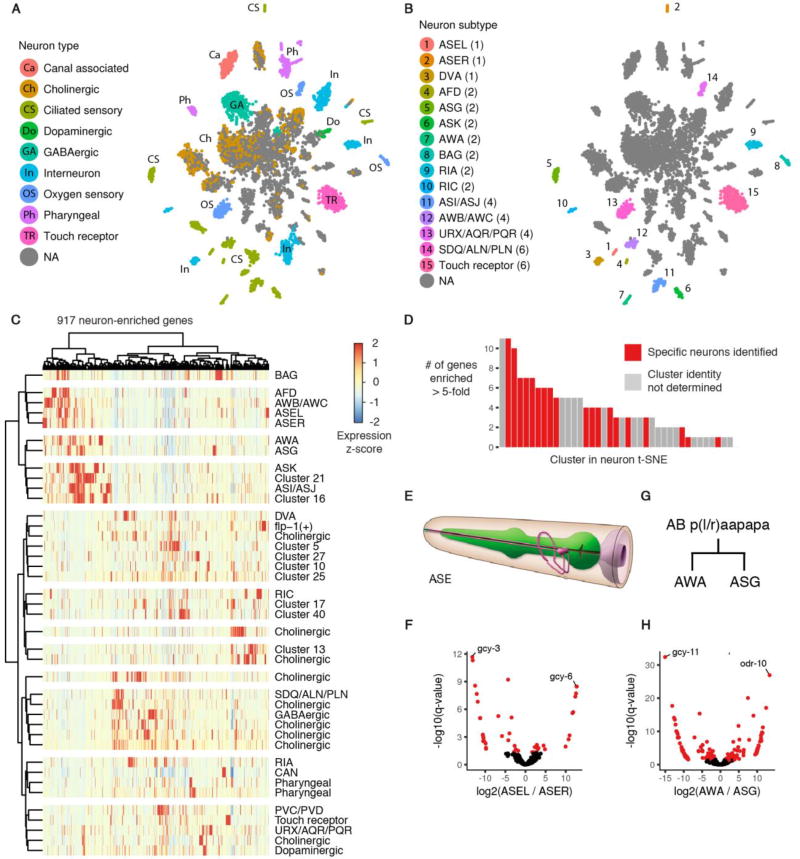
Comment in
-
Development: Transcriptomic blueprints.Nat Rev Genet. 2017 Nov;18(11):639. doi: 10.1038/nrg.2017.77. Epub 2017 Sep 18. Nat Rev Genet. 2017. PMID: 28919637 No abstract available.
References
-
- Wills QF, et al. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat. Biotechnol. 2013;31:748–752. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
