

Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells
- PMID: 28821609
- PMCID: PMC5641874
- DOI: 10.1074/jbc.M117.792838
Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells
Abstract
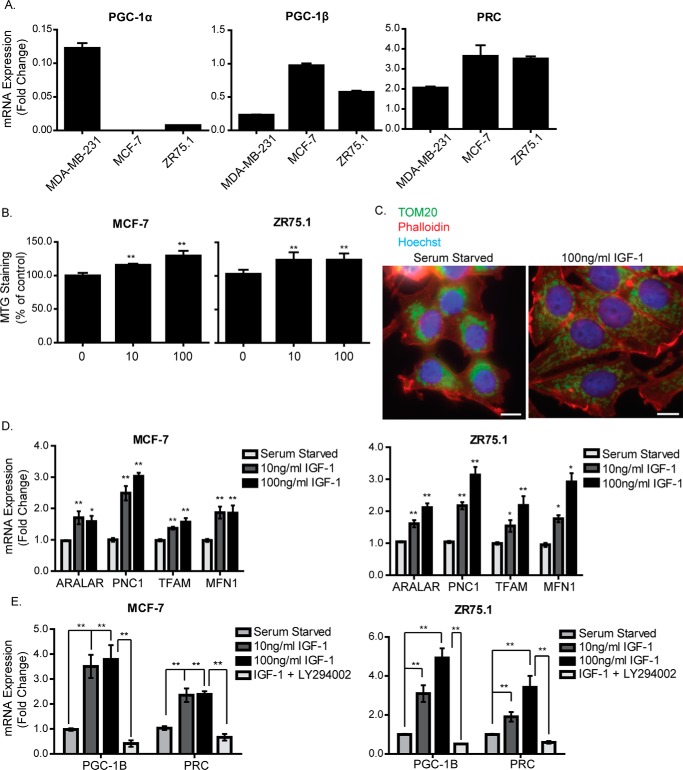
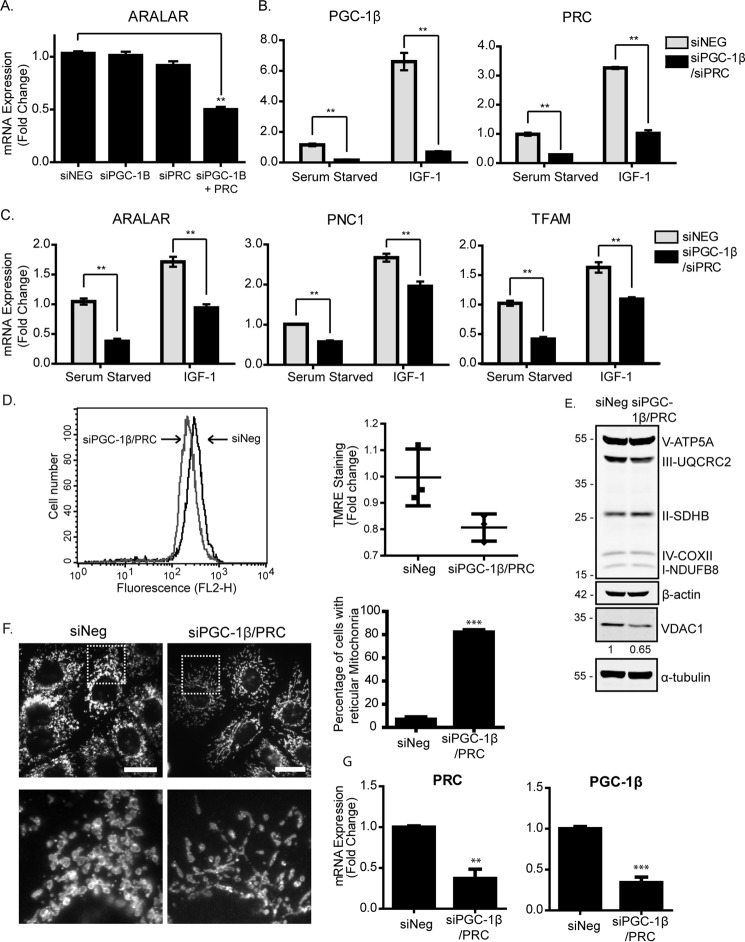
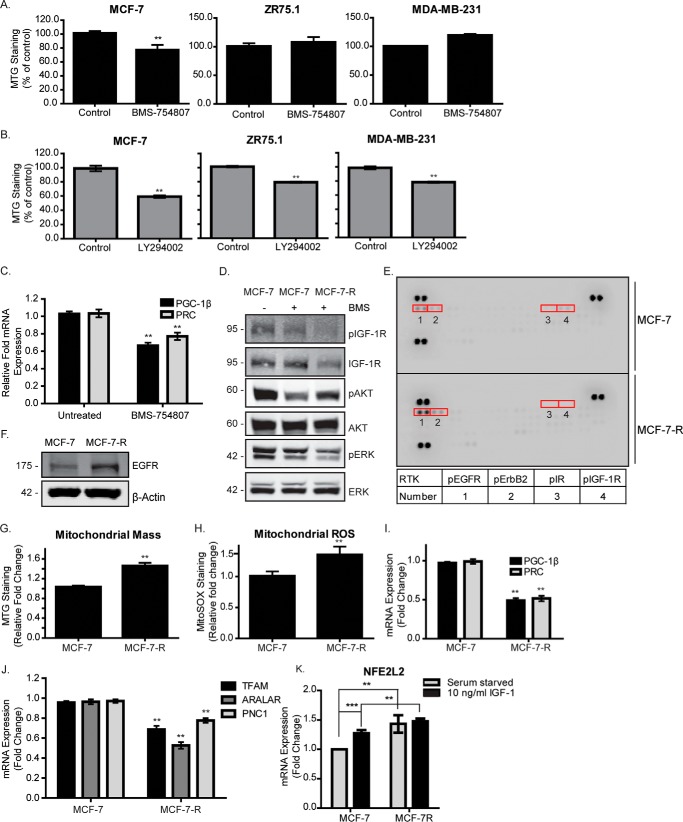
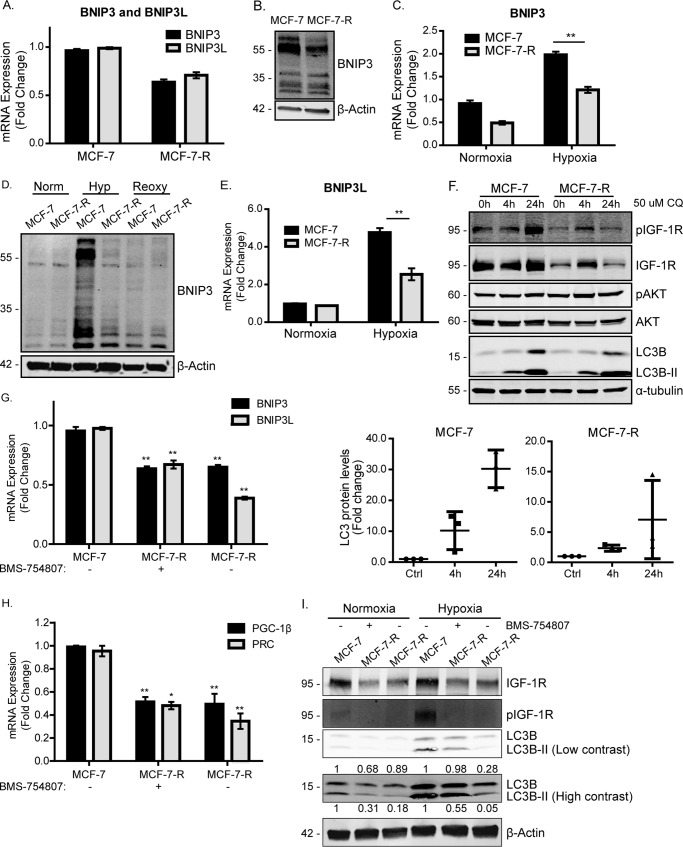
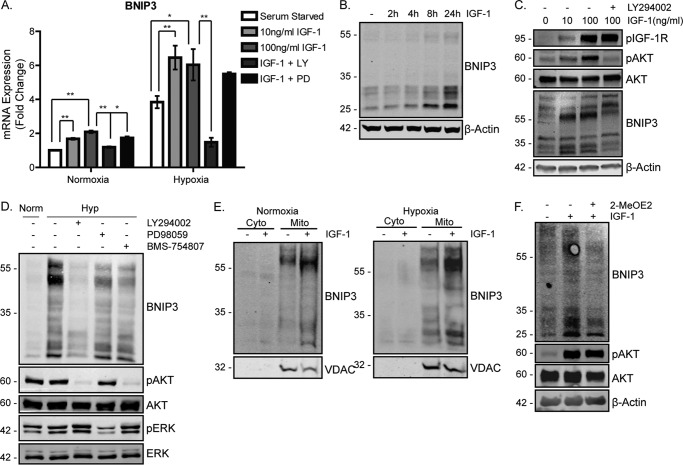
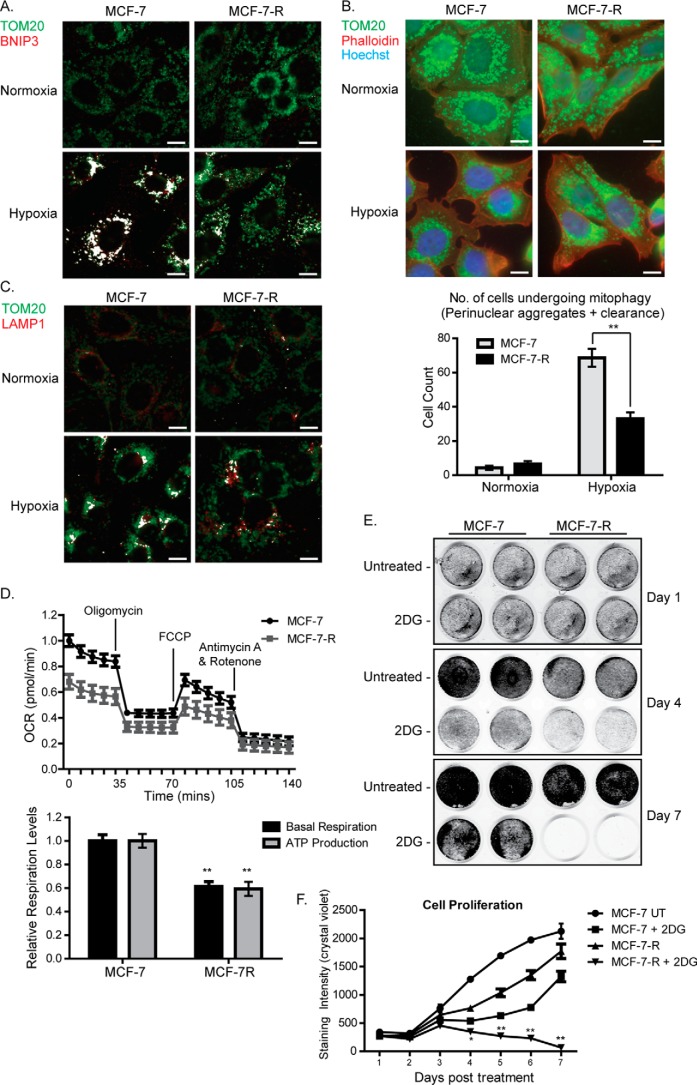
Mitochondrial activity and metabolic reprogramming influence the phenotype of cancer cells and resistance to targeted therapy. We previously established that an insulin-like growth factor 1 (IGF-1)-inducible mitochondrial UTP carrier (PNC1/SLC25A33) promotes cell growth. This prompted us to investigate whether IGF signaling is essential for mitochondrial maintenance in cancer cells and whether this contributes to therapy resistance. Here we show that IGF-1 stimulates mitochondrial biogenesis in a range of cell lines. In MCF-7 and ZR75.1 breast cancer cells, IGF-1 induces peroxisome proliferator-activated receptor γ coactivator 1β (PGC-1β) and PGC-1α-related coactivator (PRC). Suppression of PGC-1β and PRC with siRNA reverses the effects of IGF-1 and disrupts mitochondrial morphology and membrane potential. IGF-1 also induced expression of the redox regulator nuclear factor-erythroid-derived 2-like 2 (NFE2L2 alias NRF-2). Of note, MCF-7 cells with acquired resistance to an IGF-1 receptor (IGF-1R) tyrosine kinase inhibitor exhibited reduced expression of PGC-1β, PRC, and mitochondrial biogenesis. Interestingly, these cells exhibited mitochondrial dysfunction, indicated by reactive oxygen species expression, reduced expression of the mitophagy mediators BNIP3 and BNIP3L, and impaired mitophagy. In agreement with this, IGF-1 robustly induced BNIP3 accumulation in mitochondria. Other active receptor tyrosine kinases could not compensate for reduced IGF-1R activity in mitochondrial protection, and MCF-7 cells with suppressed IGF-1R activity became highly dependent on glycolysis for survival. We conclude that IGF-1 signaling is essential for sustaining cancer cell viability by stimulating both mitochondrial biogenesis and turnover through BNIP3 induction. This core mitochondrial protective signal is likely to strongly influence responses to therapy and the phenotypic evolution of cancer.
Keywords: cancer biology; cancer therapy; cell metabolism; cell signaling; cell surface receptor; drug resistance; insulin-like growth factor (IGF); mitochondria; mitophagy.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
Similar articles
-
IGF-1 Signaling Regulates Mitochondrial Remodeling during Myogenic Differentiation.Nutrients. 2022 Mar 16;14(6):1249. doi: 10.3390/nu14061249. Nutrients. 2022. PMID: 35334906 Free PMC article.
-
IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3β-Nrf2-BNIP3 Pathway.Cells. 2020 Jan 8;9(1):147. doi: 10.3390/cells9010147. Cells. 2020. PMID: 31936236 Free PMC article.
-
Decorin induces mitophagy in breast carcinoma cells via peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) and mitostatin.J Biol Chem. 2014 Feb 21;289(8):4952-68. doi: 10.1074/jbc.M113.512566. Epub 2014 Jan 8. J Biol Chem. 2014. PMID: 24403067 Free PMC article.
-
Gene methylation in gastric cancer.Clin Chim Acta. 2013 Sep 23;424:53-65. doi: 10.1016/j.cca.2013.05.002. Epub 2013 May 10. Clin Chim Acta. 2013. PMID: 23669186 Review.
-
Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX.Biochim Biophys Acta. 2015 Oct;1853(10 Pt B):2775-83. doi: 10.1016/j.bbamcr.2015.02.022. Epub 2015 Mar 6. Biochim Biophys Acta. 2015. PMID: 25753537 Review.
Cited by
-
Autophagy in Viral Development and Progression of Cancer.Front Oncol. 2021 Mar 8;11:603224. doi: 10.3389/fonc.2021.603224. eCollection 2021. Front Oncol. 2021. PMID: 33763351 Free PMC article. Review.
-
IGF-1 Signaling Regulates Mitochondrial Remodeling during Myogenic Differentiation.Nutrients. 2022 Mar 16;14(6):1249. doi: 10.3390/nu14061249. Nutrients. 2022. PMID: 35334906 Free PMC article.
-
SphK1 functions downstream of IGF-1 to modulate IGF-1-induced EMT, migration and paclitaxel resistance of A549 cells: A preliminary in vitro study.J Cancer. 2019 Jul 10;10(18):4264-4269. doi: 10.7150/jca.32646. eCollection 2019. J Cancer. 2019. PMID: 31413745 Free PMC article.
-
Pathway and Network Analyses Identify Growth Factor Signaling and MMP9 as Potential Mediators of Mitochondrial Dysfunction in Severe COVID-19.Int J Mol Sci. 2023 Jan 28;24(3):2524. doi: 10.3390/ijms24032524. Int J Mol Sci. 2023. PMID: 36768847 Free PMC article.
-
The multifaceted role of autophagy in cancer.EMBO J. 2022 Jul 4;41(13):e110031. doi: 10.15252/embj.2021110031. Epub 2022 May 10. EMBO J. 2022. PMID: 35535466 Free PMC article. Review.
References
-
- Palikaras K., Lionaki E., and Tavernarakis N. (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528 - PubMed
-
- Raimundo N. (2014) Mitochondrial pathology: stress signals from the energy factory. Trends Mol. Med. 20, 282–292 - PubMed
-
- Marín de Mas I., Aguilar E., Jayaraman A., Polat I. H., Martín-Bernabé A., Bharat R., Foguet C., Milà E., Papp B., Centelles J. J., and Cascante M. (2014) Cancer cell metabolism as new targets for novel designed therapies. Future Med. Chem. 6, 1791–1810 - PubMed
-
- Moreno-Sánchez R., Marín-Hernández A., Saavedra E., Pardo J. P., Ralph S. J., and Rodríguez-Enríquez S. (2014) Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int. J. Biochem. Cell Biol. 50, 10–23 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
