

Distinct structural mechanisms determine substrate affinity and kinase activity of protein kinase Cα
- PMID: 28821615
- PMCID: PMC5625059
- DOI: 10.1074/jbc.M117.804781
Distinct structural mechanisms determine substrate affinity and kinase activity of protein kinase Cα
Abstract
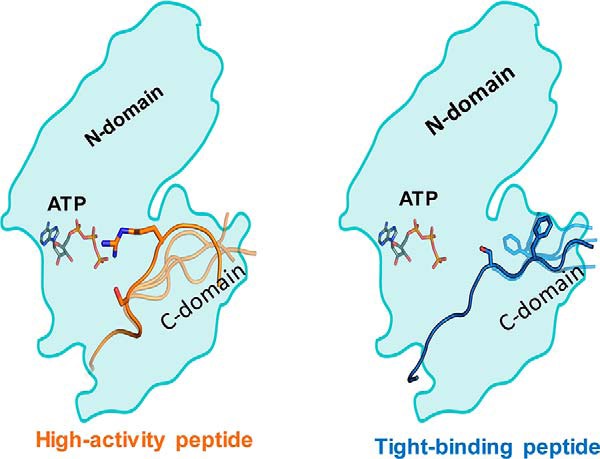
Protein kinase Cα (PKCα) belongs to the family of AGC kinases that phosphorylate multiple peptide substrates. Although the consensus sequence motif has been identified and used to explain substrate specificity for PKCα, it does not inform the structural basis of substrate-binding and kinase activity for diverse substrates phosphorylated by this kinase. The transient, dynamic, and unstructured nature of this protein-protein interaction has limited structural mapping of kinase-substrate interfaces. Here, using multiscale MD simulation-based predictions and FRET sensor-based experiments, we investigated the conformational dynamics of the kinase-substrate interface. We found that the binding strength of the kinase-substrate interaction is primarily determined by long-range columbic interactions between basic (Arg/Lys) residues located N-terminally to the phosphorylated Ser/Thr residues in the substrate and by an acidic patch in the kinase catalytic domain. Kinase activity stemmed from conformational flexibility in the region C-terminal to the phosphorylated Ser/Thr residues. Flexibility of the substrate-kinase interaction enabled an Arg/Lys two to three amino acids C-terminal to the phosphorylated Ser/Thr to prime a catalytically active conformation, facilitating phosphoryl transfer to the substrate. The structural mechanisms determining substrate binding and catalytic activity formed the basis of diverse binding affinities and kinase activities of PKCα for 14 substrates with varying degrees of sequence conservation. Our findings provide insight into the dynamic properties of the kinase-substrate interaction that govern substrate binding and turnover. Moreover, this study establishes a modeling and experimental method to elucidate the structural dynamics underlying substrate selectivity among eukaryotic kinases.
Keywords: GNEIMO; binding affinity; conformational change; fluorescence resonance energy transfer (FRET); molecular dynamics; multiscale molecular dynamics; peptide interaction; protein conformation; protein kinase C (PKC); protein phosphorylation.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
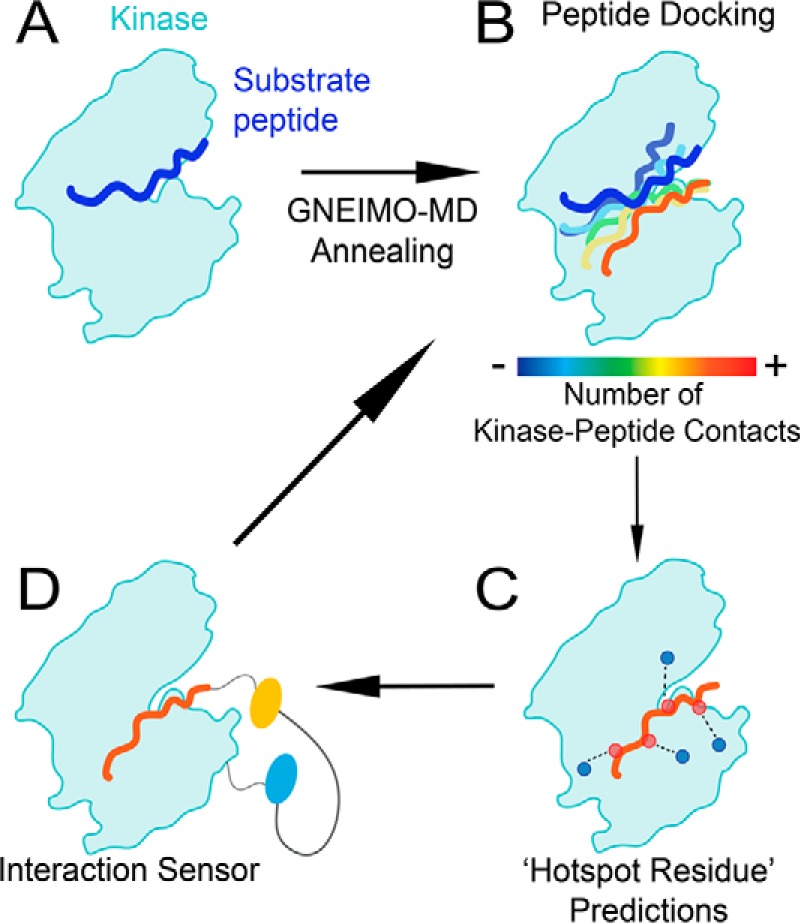
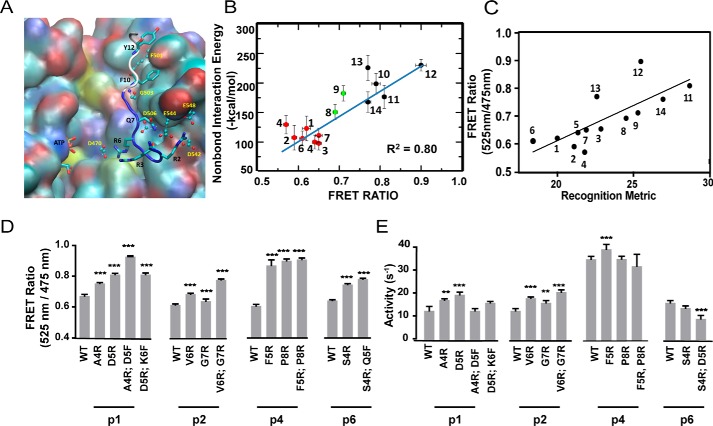
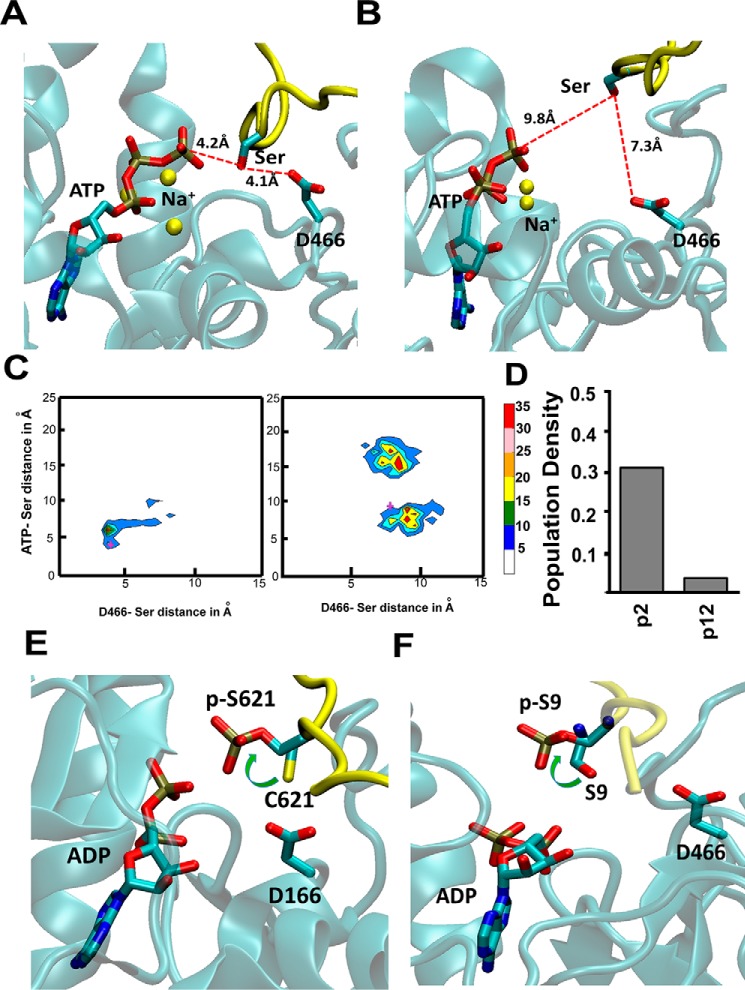
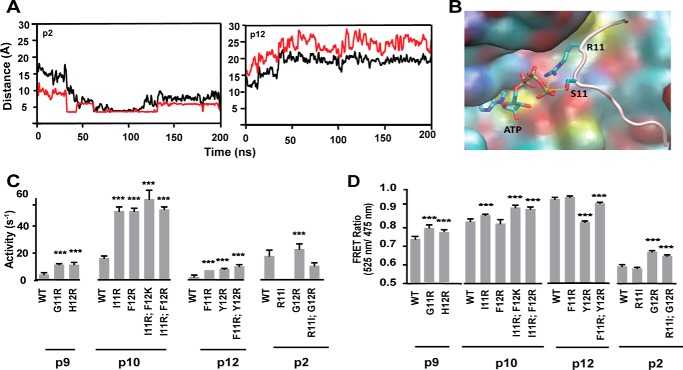
References
-
- Endicott J. A., Noble M. E., and Johnson L. N. (2012) The structural basis of control of eukaryotic protein kinases. Annu. Rev. Biochem. 81, 587–613 - PubMed
-
- de Oliveira P. S., Ferraz F. A., Pena D. A., Pramio D. T., Morais F. A., and Schechtman D. (2016) Revisting protein kinase-substrate interactions: towards therapeutic development. Sci. Signal. 9, re3. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
