

Neuronal Depolarization Drives Increased Dopamine Synaptic Vesicle Loading via VGLUT
- PMID: 28823729
- PMCID: PMC5760215
- DOI: 10.1016/j.neuron.2017.07.038
Neuronal Depolarization Drives Increased Dopamine Synaptic Vesicle Loading via VGLUT
Abstract
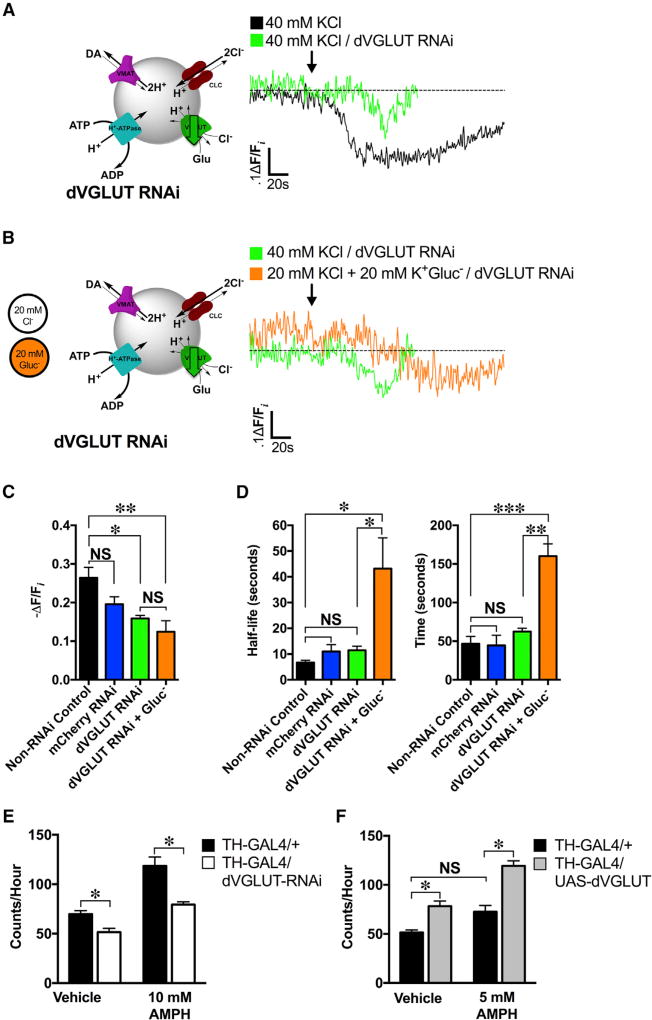
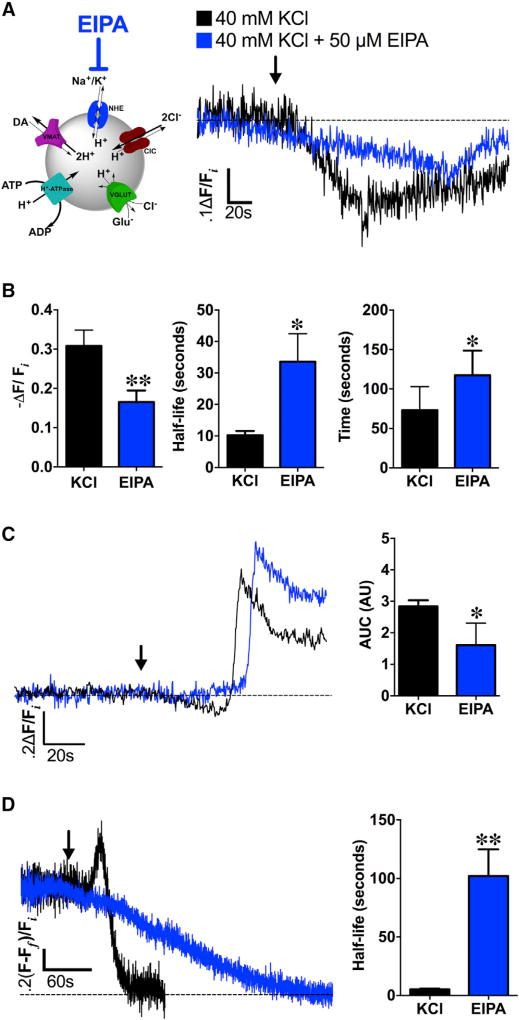
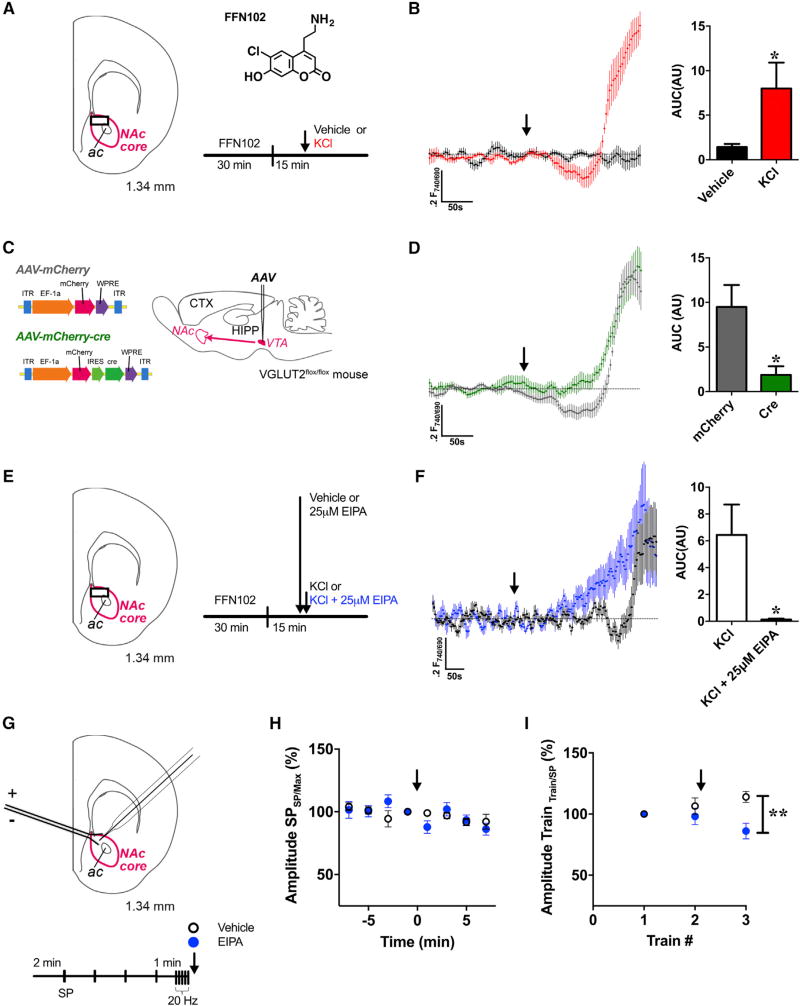
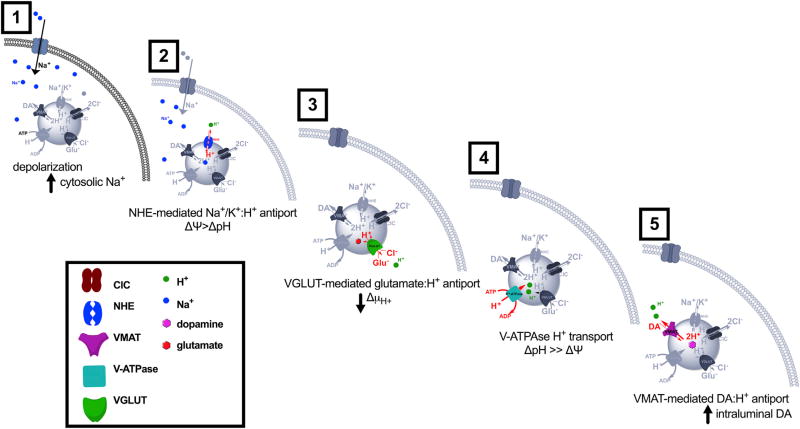
The ability of presynaptic dopamine terminals to tune neurotransmitter release to meet the demands of neuronal activity is critical to neurotransmission. Although vesicle content has been assumed to be static, in vitro data increasingly suggest that cell activity modulates vesicle content. Here, we use a coordinated genetic, pharmacological, and imaging approach in Drosophila to study the presynaptic machinery responsible for these vesicular processes in vivo. We show that cell depolarization increases synaptic vesicle dopamine content prior to release via vesicular hyperacidification. This depolarization-induced hyperacidification is mediated by the vesicular glutamate transporter (VGLUT). Remarkably, both depolarization-induced dopamine vesicle hyperacidification and its dependence on VGLUT2 are seen in ventral midbrain dopamine neurons in the mouse. Together, these data suggest that in response to depolarization, dopamine vesicles utilize a cascade of vesicular transporters to dynamically increase the vesicular pH gradient, thereby increasing dopamine vesicle content.
Keywords: VGLUT2; depolarization; dopamine; glutamate; neurotransmission; pH; presynaptic; synaptic vesicle; vesicle content.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
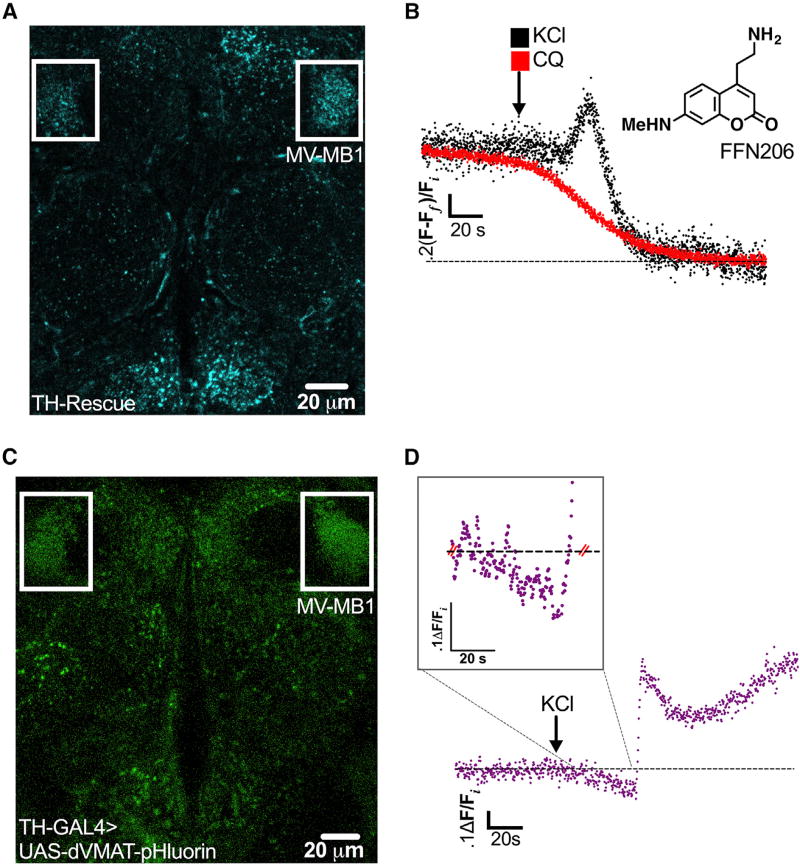
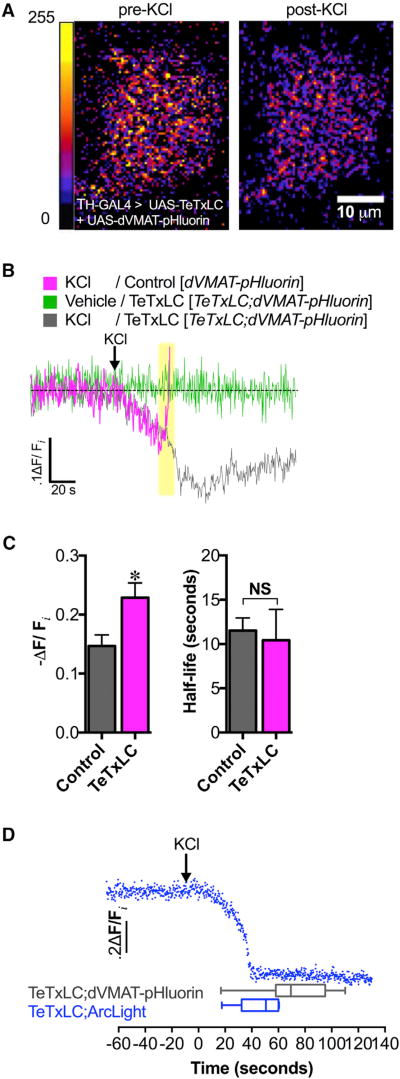
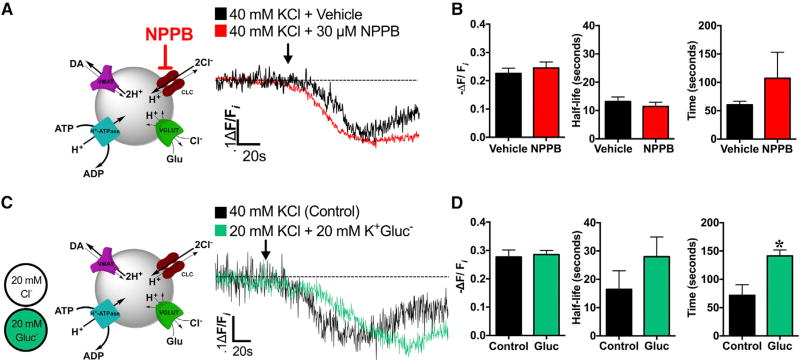
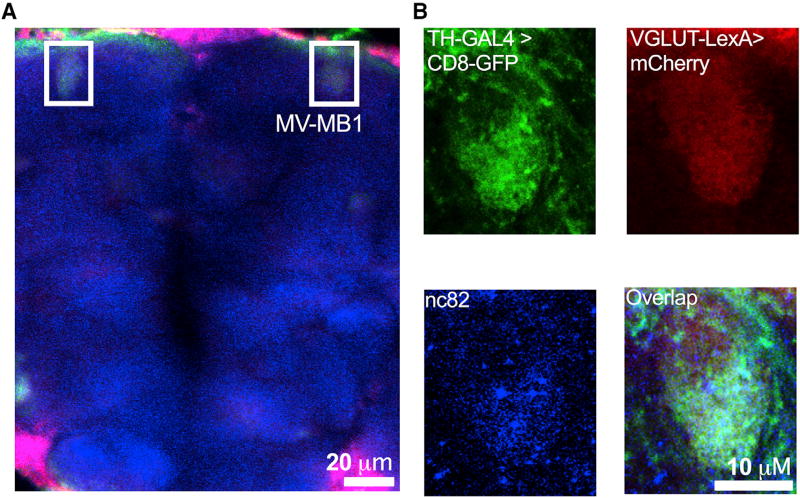
Comment in
-
Depolarization-induced synaptic vesicle hyperacidification: A new path regarding dopamine release.Mov Disord. 2017 Dec;32(12):1684. doi: 10.1002/mds.27210. Epub 2017 Nov 9. Mov Disord. 2017. PMID: 29119595 No abstract available.
References
MeSH terms
Substances
Grants and funding
- P01 DA010154/DA/NIDA NIH HHS/United States
- T32 GM007628/GM/NIGMS NIH HHS/United States
- R01 MH108186/MH/NIMH NIH HHS/United States
- R01 ES015747/ES/NIEHS NIH HHS/United States
- R01 MH086545/MH/NIMH NIH HHS/United States
- R21 NS075572/NS/NINDS NIH HHS/United States
- P50 AG008702/AG/NIA NIH HHS/United States
- R01 MH107390/MH/NIMH NIH HHS/United States
- R01 NS075222/NS/NINDS NIH HHS/United States
- K05 DA022413/DA/NIDA NIH HHS/United States
- R01 DA038966/DA/NIDA NIH HHS/United States
- R01 MH076900/MH/NIMH NIH HHS/United States
- P01 DA012408/DA/NIDA NIH HHS/United States
- P01 ES016732/ES/NIEHS NIH HHS/United States
- K08 DA031241/DA/NIDA NIH HHS/United States
- R01 DA007418/DA/NIDA NIH HHS/United States
- F31 MH114316/MH/NIMH NIH HHS/United States
- R21 DA040443/DA/NIDA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
