

The Enigmatic Role of C9ORF72 in Autophagy
- PMID: 28824365
- PMCID: PMC5541066
- DOI: 10.3389/fnins.2017.00442
The Enigmatic Role of C9ORF72 in Autophagy
Abstract
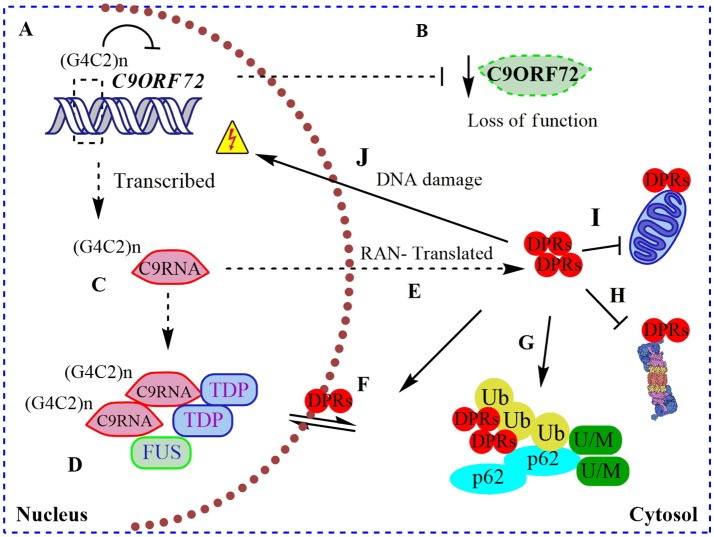
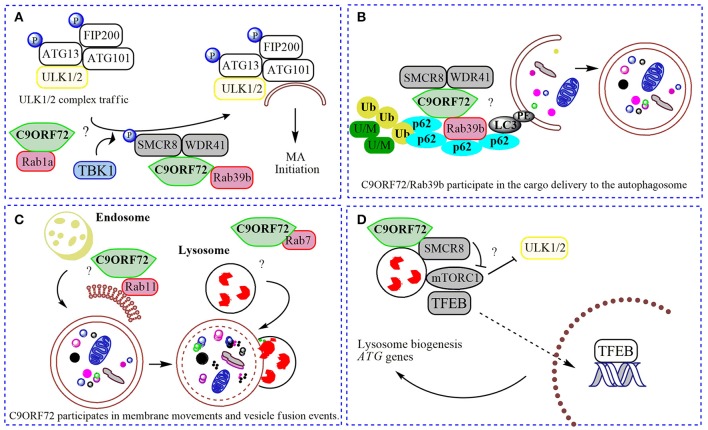
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by the loss of motor neurons resulting in a progressive and irreversible muscular paralysis. Advances in large-scale genetics and genomics have revealed intronic hexanucleotide repeat expansions in the gene encoding C9ORF72 as a main genetic cause of ALS and frontotemporal dementia (FTD), the second most common cause of early-onset dementia after Alzheimer's disease. Novel insights regarding the underlying pathogenic mechanisms of C9ORF72 seem to suggest a synergy of loss and gain of toxic function during disease. C9ORF72, thus far, has been found to be involved in homeostatic cellular pathways, such as actin dynamics, regulation of membrane trafficking, and macroautophagy. All these pathways have been found compromised in the pathogenesis of ALS. In this review, we aim to summarize recent findings on the function of C9ORF72, particularly in the macroautophagy pathway, hinting at a requirement to maintain the fine balance of macroautophagy to prevent neurodegeneration.
Keywords: ALS; C9ORF72; FTD; autophagy; macroautophagy.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous