

A Normalization-Free and Nonparametric Method Sharpens Large-Scale Transcriptome Analysis and Reveals Common Gene Alteration Patterns in Cancers
- PMID: 28824723
- PMCID: PMC5562223
- DOI: 10.7150/thno.19425
A Normalization-Free and Nonparametric Method Sharpens Large-Scale Transcriptome Analysis and Reveals Common Gene Alteration Patterns in Cancers
Abstract
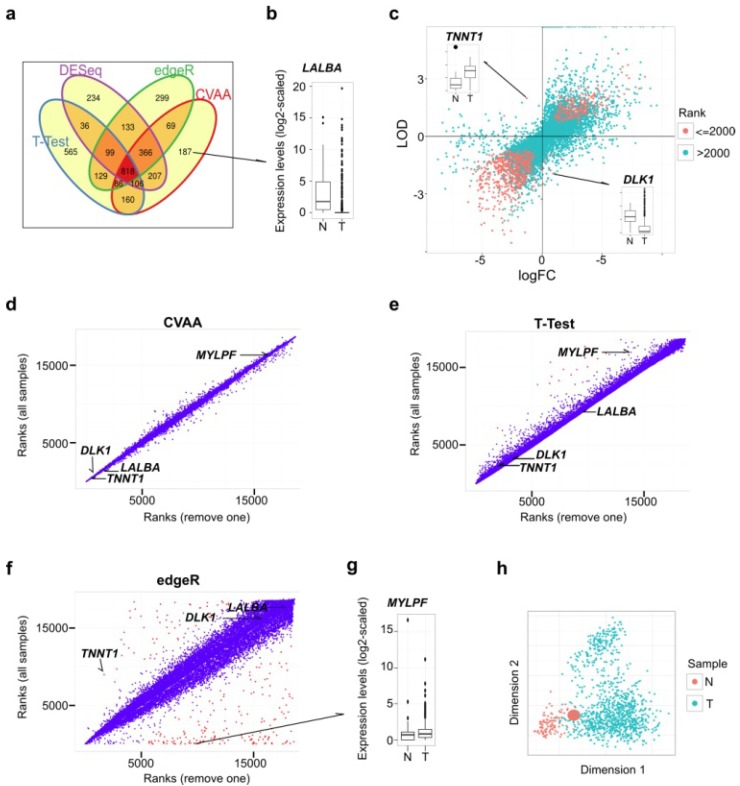
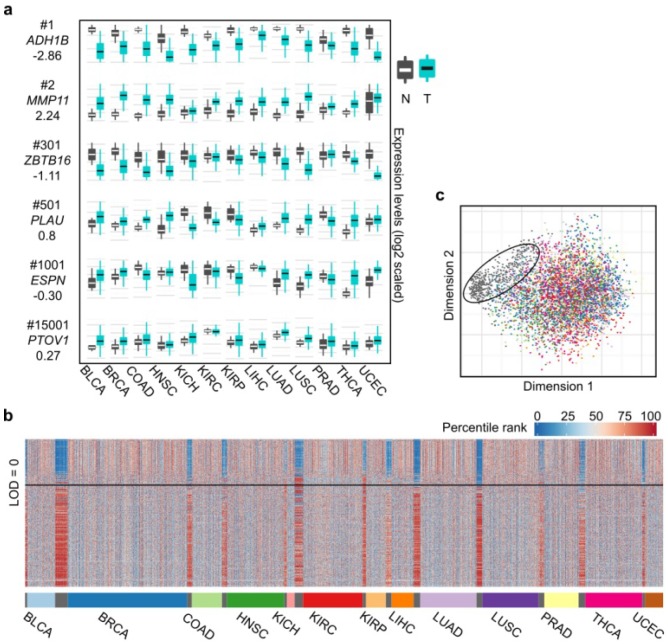
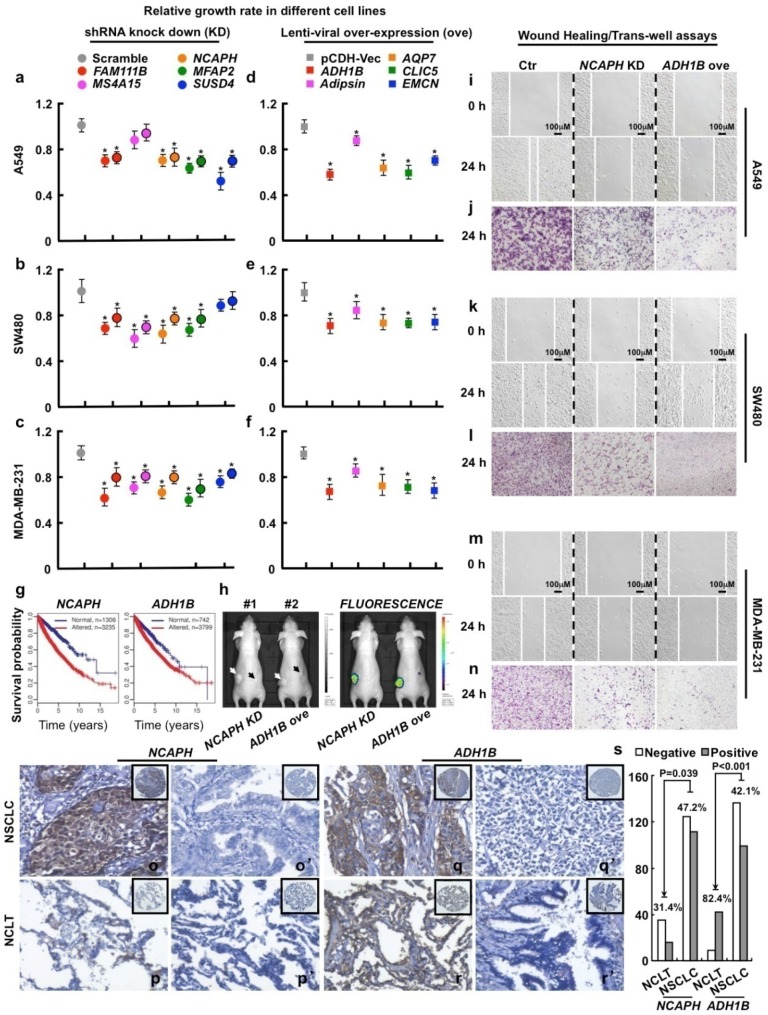
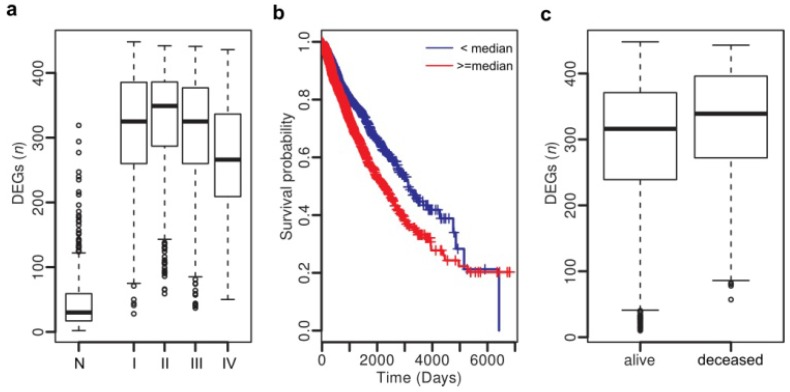
Heterogeneity in transcriptional data hampers the identification of differentially expressed genes (DEGs) and understanding of cancer, essentially because current methods rely on cross-sample normalization and/or distribution assumption-both sensitive to heterogeneous values. Here, we developed a new method, Cross-Value Association Analysis (CVAA), which overcomes the limitation and is more robust to heterogeneous data than the other methods. Applying CVAA to a more complex pan-cancer dataset containing 5,540 transcriptomes discovered numerous new DEGs and many previously rarely explored pathways/processes; some of them were validated, both in vitro and in vivo, to be crucial in tumorigenesis, e.g., alcohol metabolism (ADH1B), chromosome remodeling (NCAPH) and complement system (Adipsin). Together, we present a sharper tool to navigate large-scale expression data and gain new mechanistic insights into tumorigenesis.
Keywords: Cross-Value Association Analysis; heterogeneity.; normalization-free; pan-cancer; transcriptome.
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
