

Evolutionarily Conserved Principles Predict 3D Chromatin Organization
- PMID: 28826674
- PMCID: PMC5591081
- DOI: 10.1016/j.molcel.2017.07.022
Evolutionarily Conserved Principles Predict 3D Chromatin Organization
Abstract
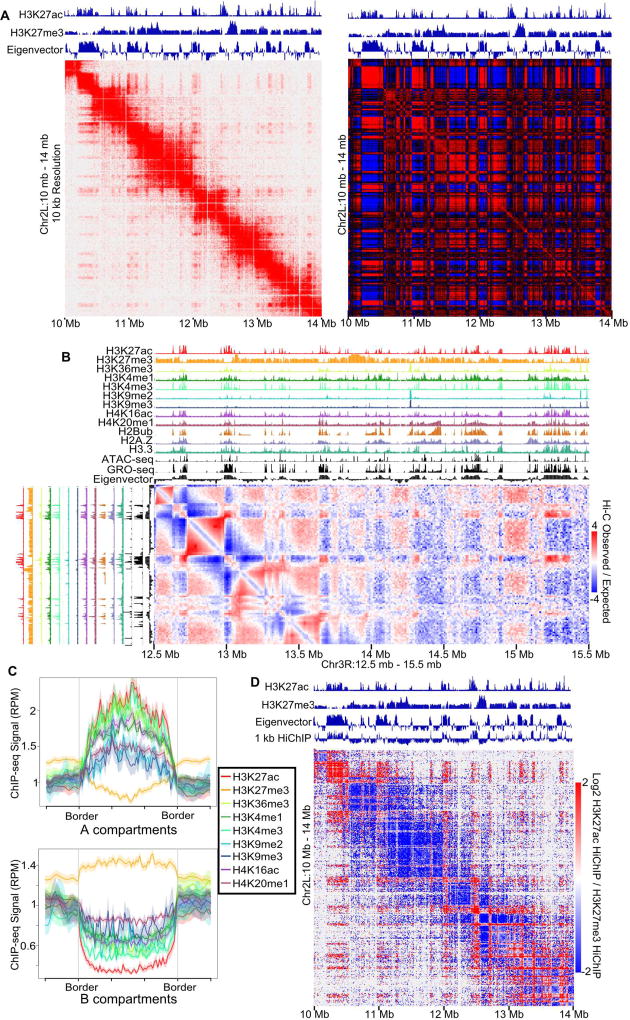
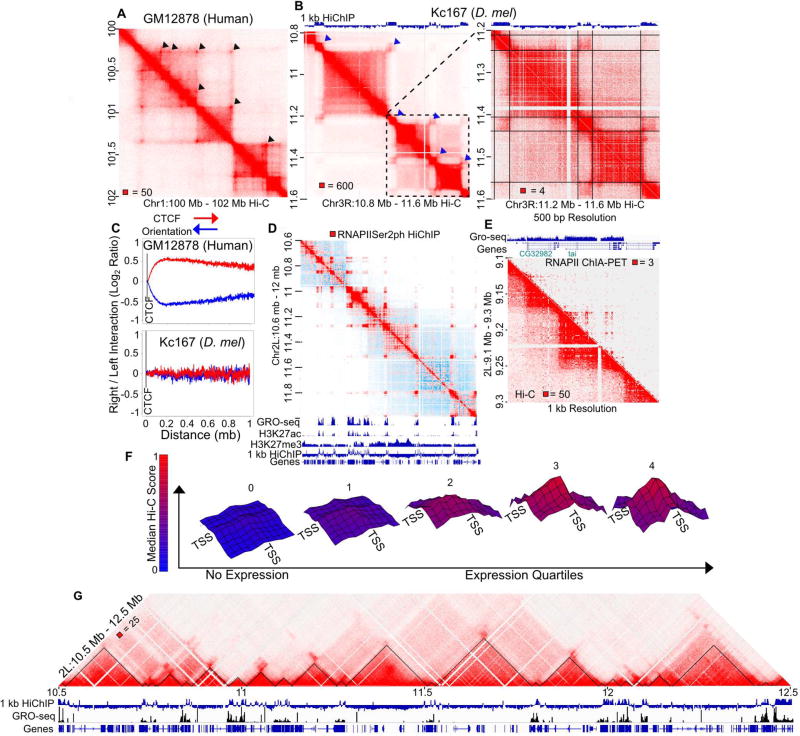
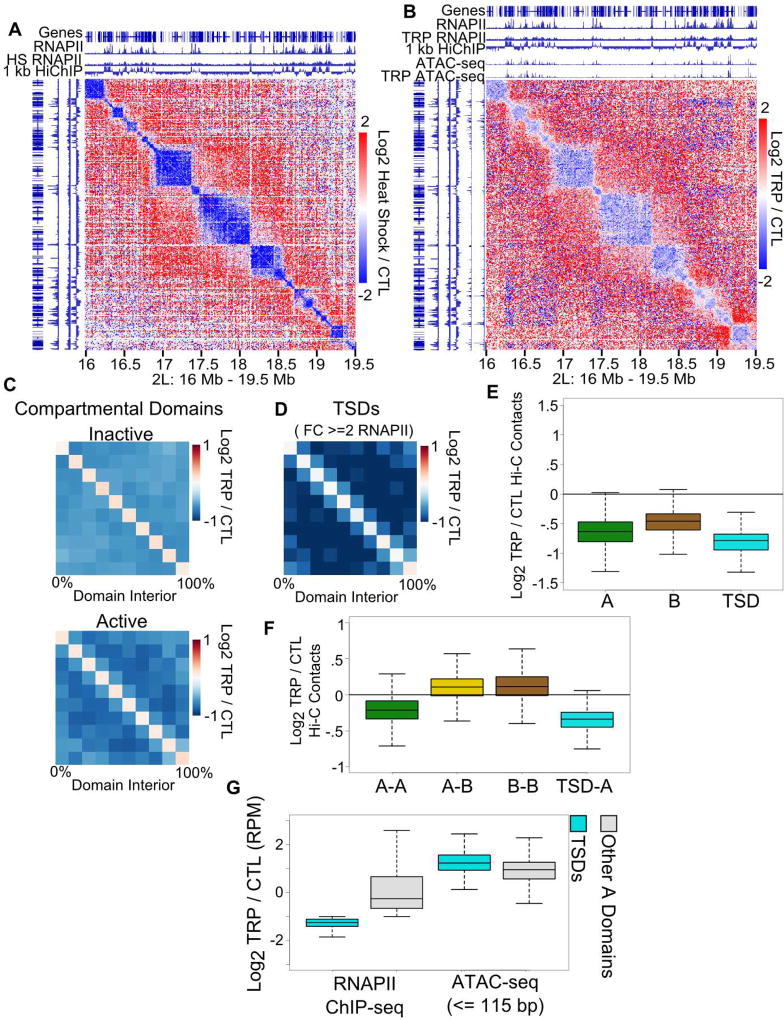
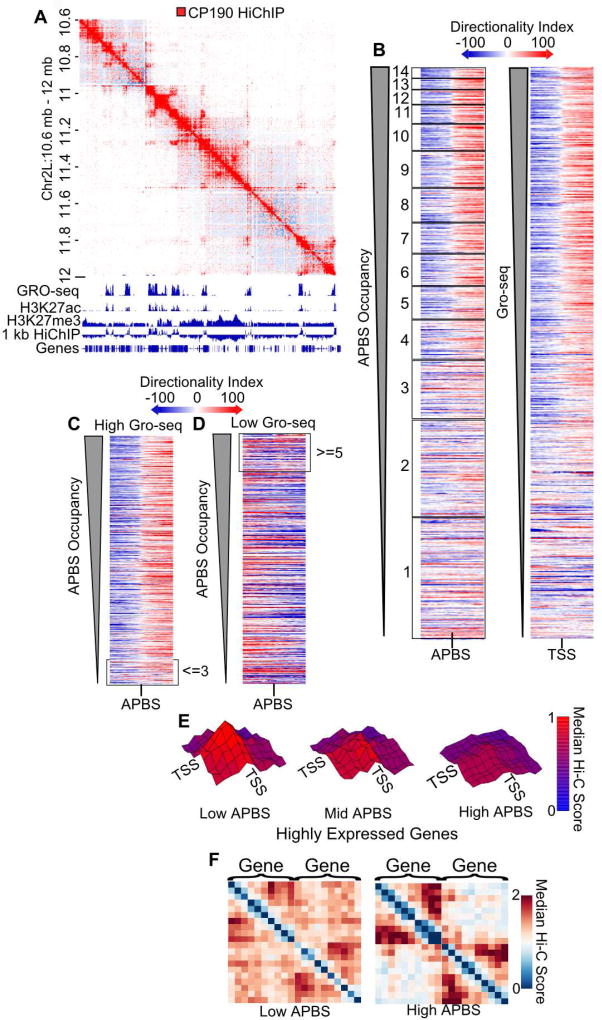
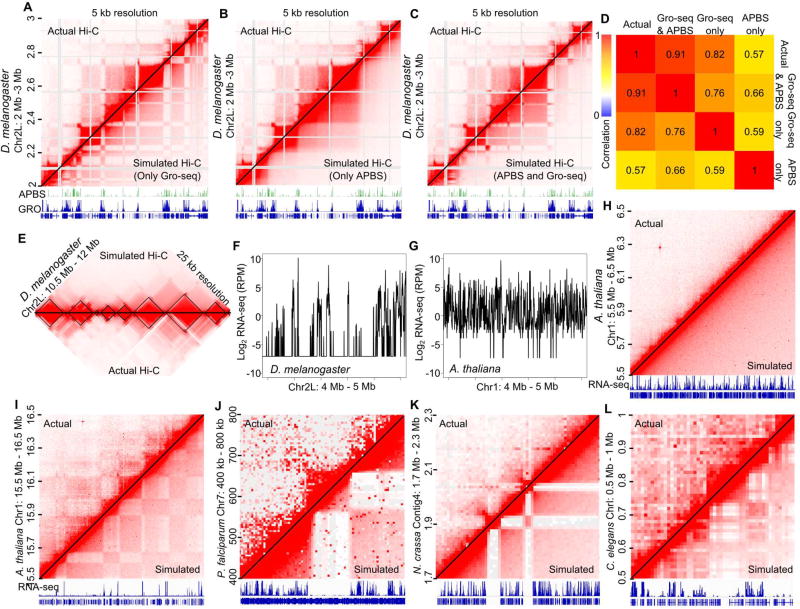
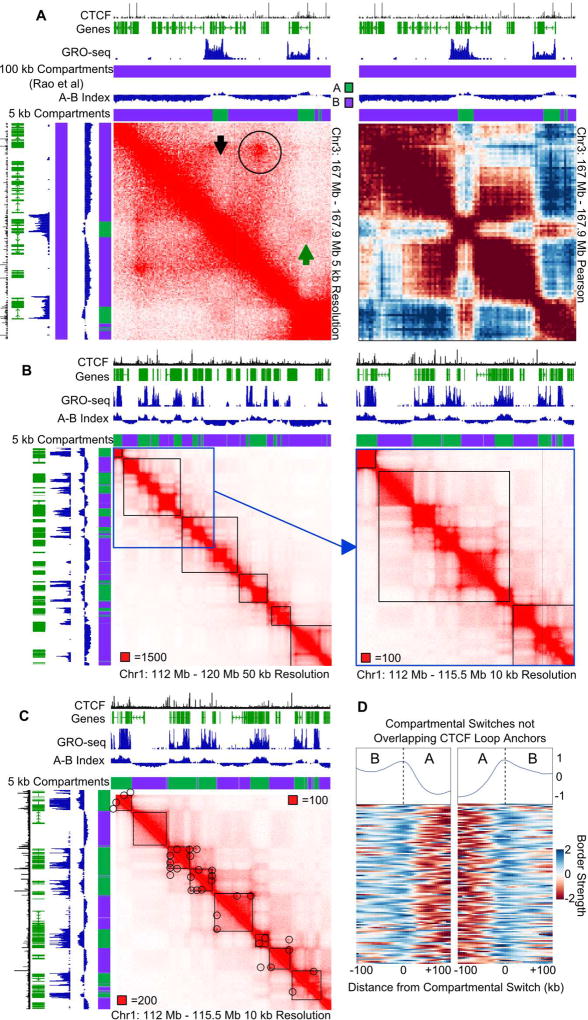
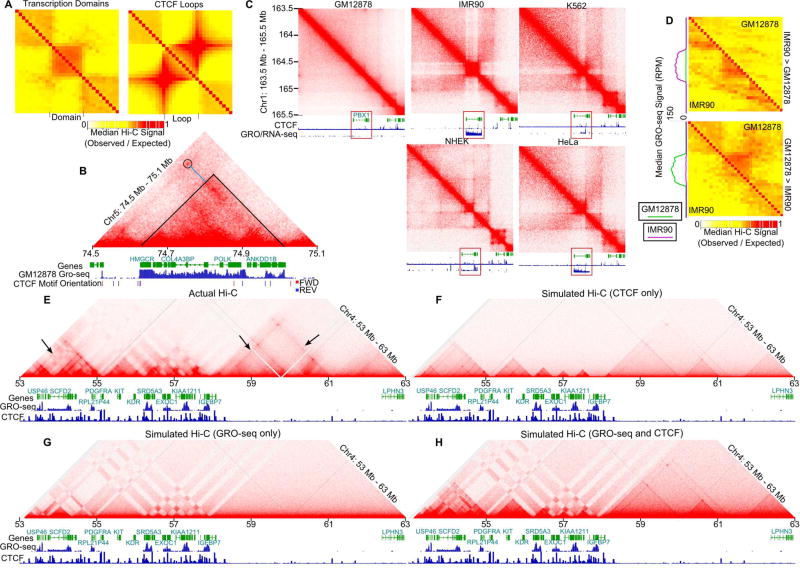
Topologically associating domains (TADs), CTCF loop domains, and A/B compartments have been identified as important structural and functional components of 3D chromatin organization, yet the relationship between these features is not well understood. Using high-resolution Hi-C and HiChIP, we show that Drosophila chromatin is organized into domains we term compartmental domains that correspond precisely with A/B compartments at high resolution. We find that transcriptional state is a major predictor of Hi-C contact maps in several eukaryotes tested, including C. elegans and A. thaliana. Architectural proteins insulate compartmental domains by reducing interaction frequencies between neighboring regions in Drosophila, but CTCF loops do not play a distinct role in this organism. In mammals, compartmental domains exist alongside CTCF loop domains to form topological domains. The results suggest that compartmental domains are responsible for domain structure in all eukaryotes, with CTCF playing an important role in domain formation in mammals.
Keywords: CTCF; Hi-C; TAD; compartment; epigenetics; insulator; loop; transcription.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Andersson R, Refsing Andersen P, Valen E, Core LJ, Bornholdt J, Boyd M, Heick Jensen T, Sandelin A. Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat. Commun. 2014;5:5336. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
