

Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry
- PMID: 28827567
- PMCID: PMC5566333
- DOI: 10.1038/s41467-017-00249-5
Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry
Abstract
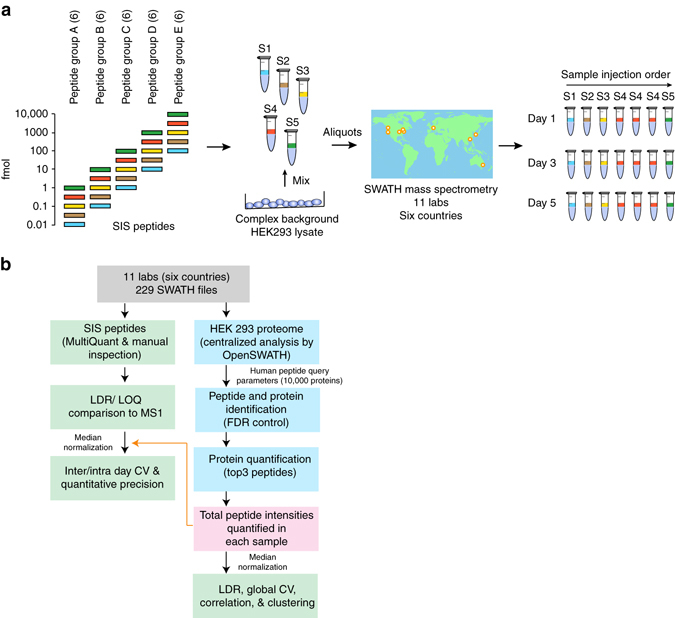
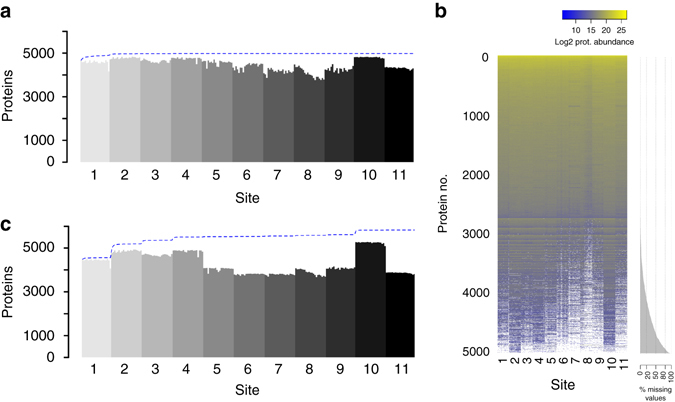
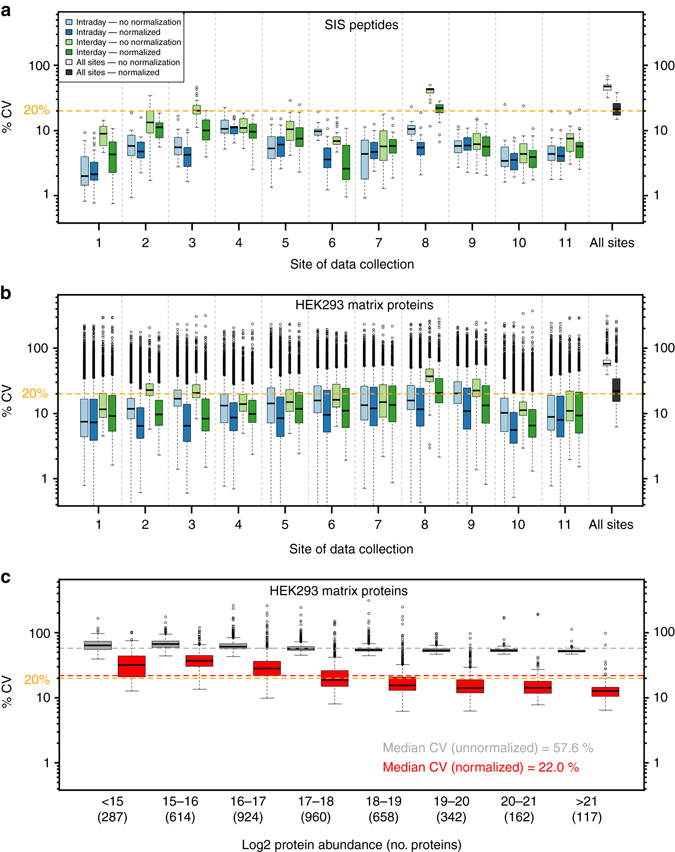
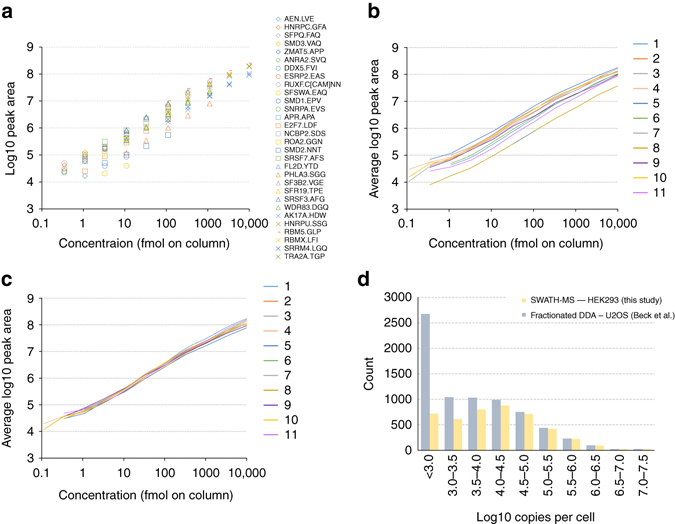
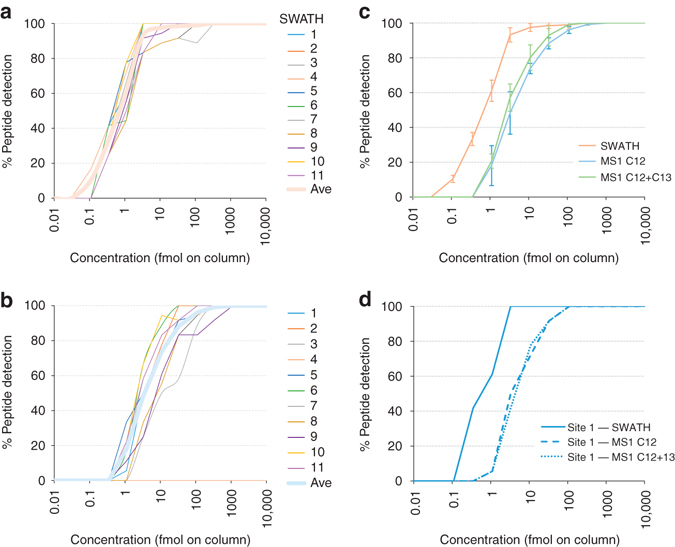
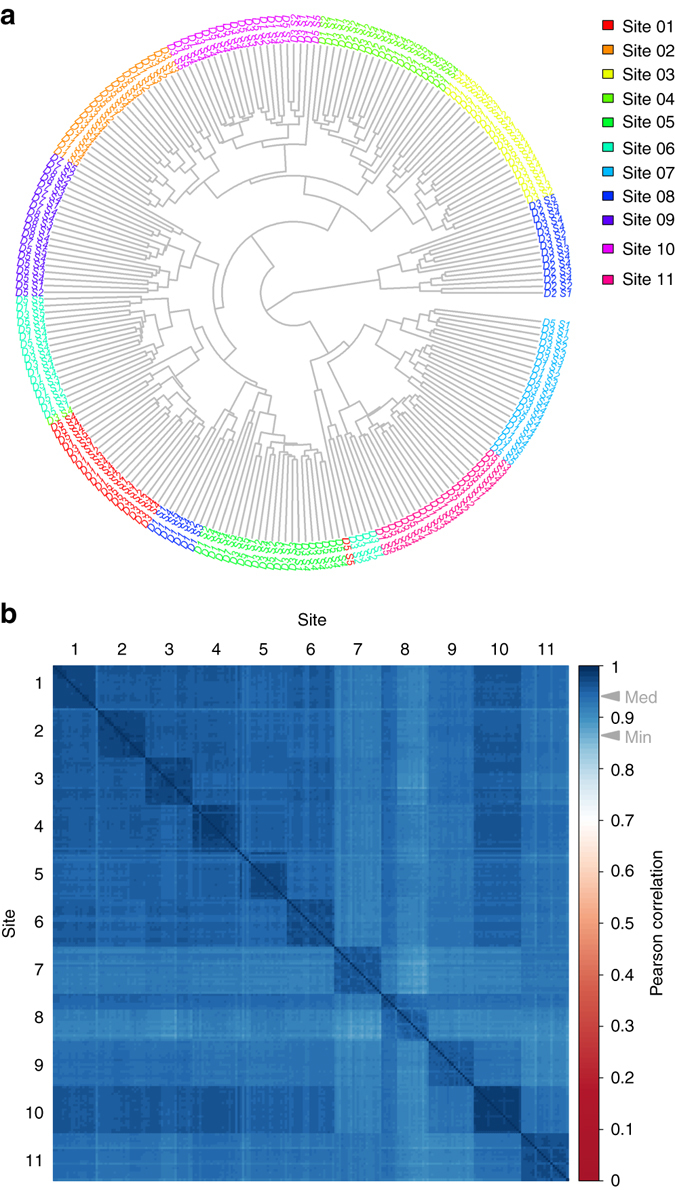
Quantitative proteomics employing mass spectrometry is an indispensable tool in life science research. Targeted proteomics has emerged as a powerful approach for reproducible quantification but is limited in the number of proteins quantified. SWATH-mass spectrometry consists of data-independent acquisition and a targeted data analysis strategy that aims to maintain the favorable quantitative characteristics (accuracy, sensitivity, and selectivity) of targeted proteomics at large scale. While previous SWATH-mass spectrometry studies have shown high intra-lab reproducibility, this has not been evaluated between labs. In this multi-laboratory evaluation study including 11 sites worldwide, we demonstrate that using SWATH-mass spectrometry data acquisition we can consistently detect and reproducibly quantify >4000 proteins from HEK293 cells. Using synthetic peptide dilution series, we show that the sensitivity, dynamic range and reproducibility established with SWATH-mass spectrometry are uniformly achieved. This study demonstrates that the acquisition of reproducible quantitative proteomics data by multiple labs is achievable, and broadly serves to increase confidence in SWATH-mass spectrometry data acquisition as a reproducible method for large-scale protein quantification.SWATH-mass spectrometry consists of a data-independent acquisition and a targeted data analysis strategy that aims to maintain the favorable quantitative characteristics on the scale of thousands of proteins. Here, using data generated by eleven groups worldwide, the authors show that SWATH-MS is capable of generating highly reproducible data across different laboratories.
Conflict of interest statement
C.H. is an employee of SCIEX, which operates in the field covered by the article. R.A. holds shares of Biognosys AG which operates in the field covered by the article. The remaining authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
