

Polymodal allosteric regulation of Type 1 Serine/Threonine Kinase Receptors via a conserved electrostatic lock
- PMID: 28827795
- PMCID: PMC5578689
- DOI: 10.1371/journal.pcbi.1005711
Polymodal allosteric regulation of Type 1 Serine/Threonine Kinase Receptors via a conserved electrostatic lock
Abstract
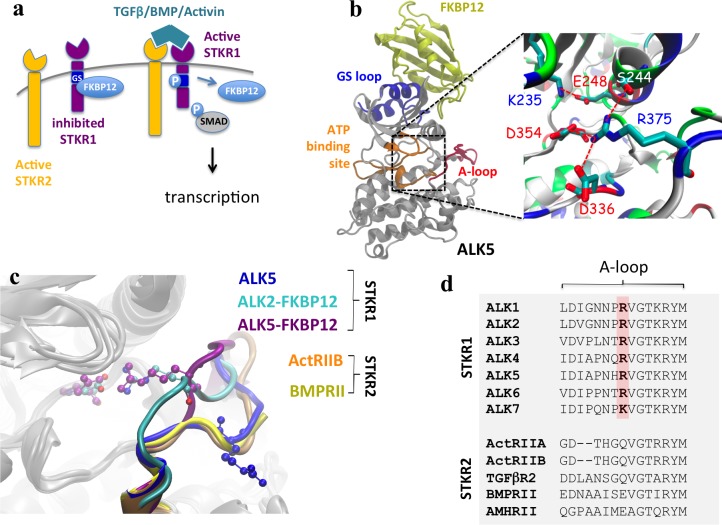
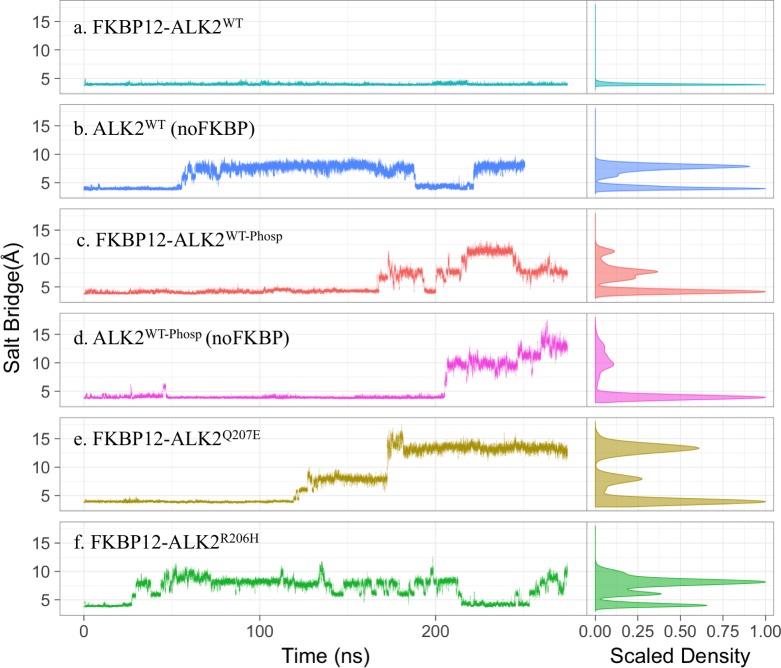
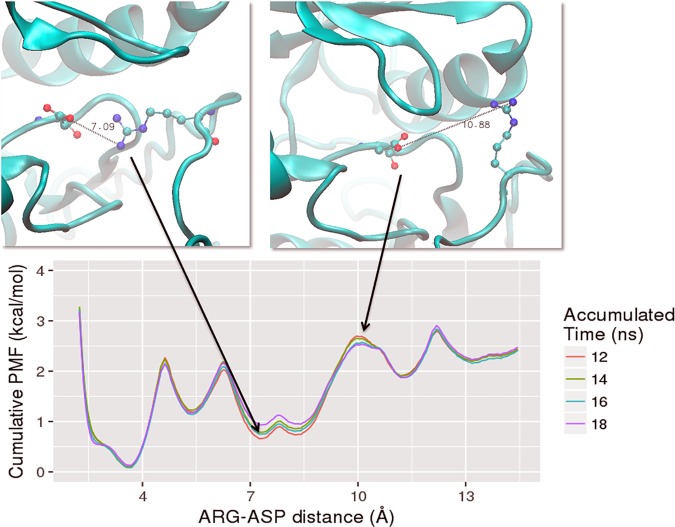
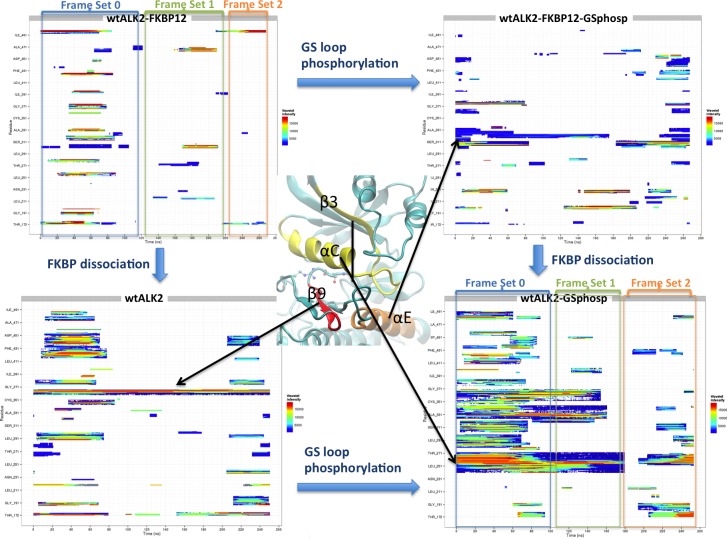
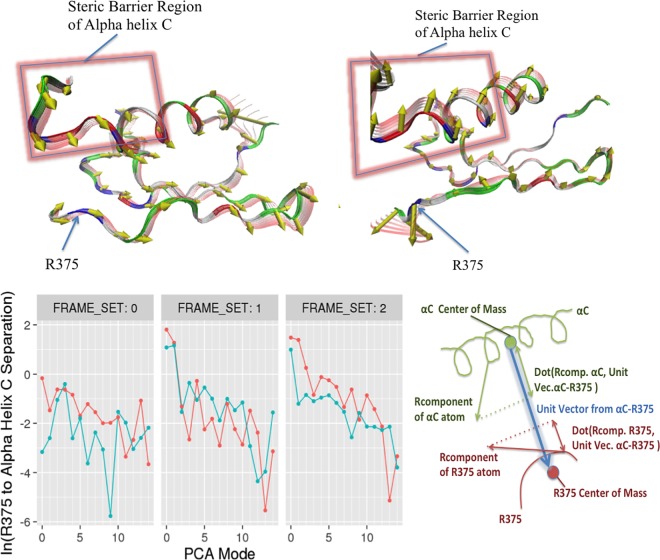
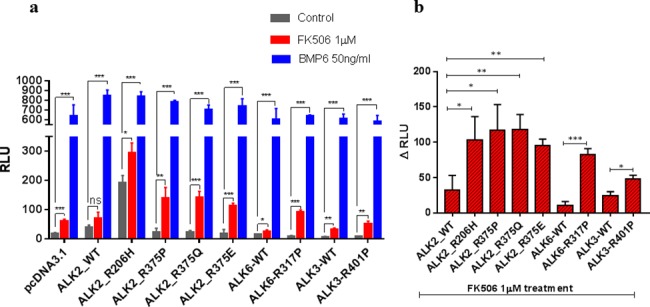
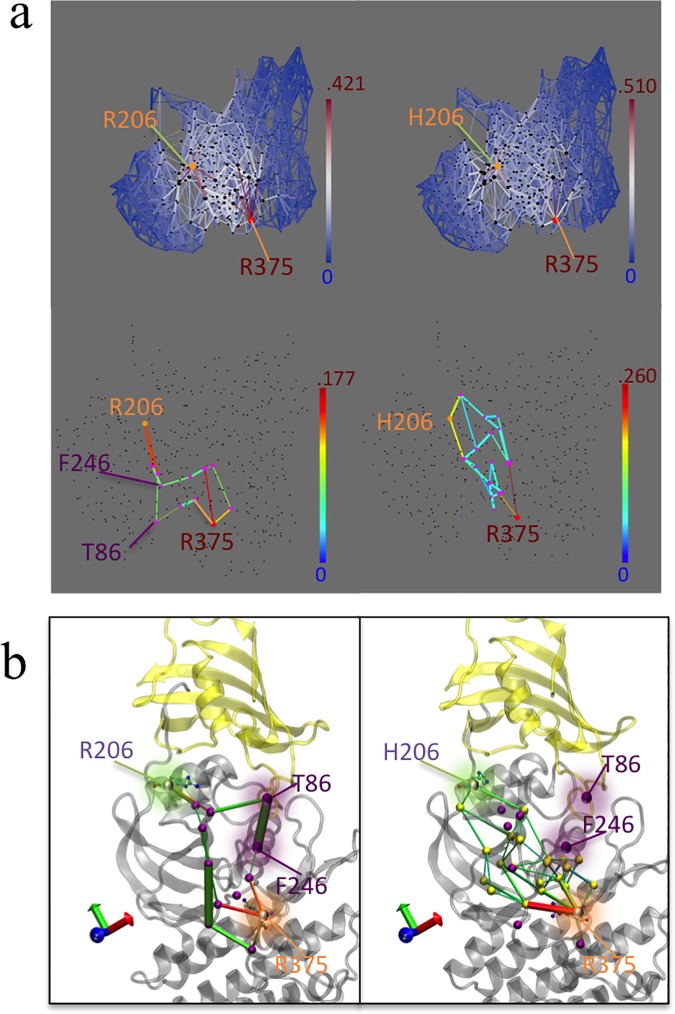
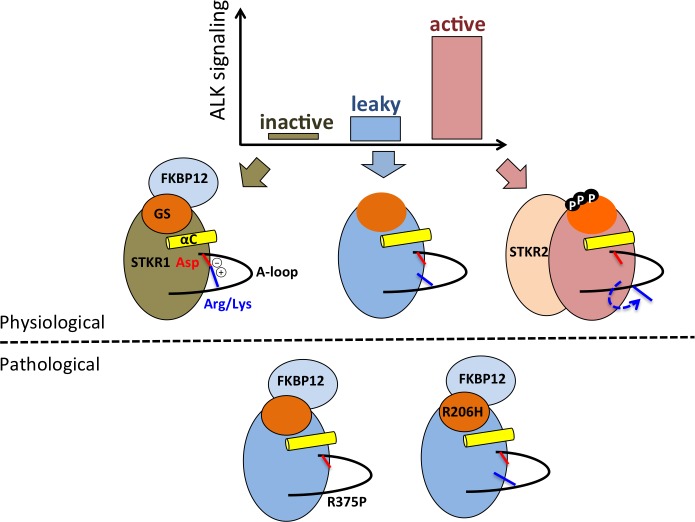
Type 1 Serine/Threonine Kinase Receptors (STKR1) transduce a wide spectrum of biological signals mediated by TGF-β superfamily members. The STKR1 activity is tightly controlled by their regulatory glycine-serine rich (GS) domain adjacent to the kinase domain. Despite decades of studies, it remains unknown how physiological or pathological GS domain modifications are coupled to STKR1 kinase activity. Here, by performing molecular dynamics simulations and free energy calculation of Activin-Like Kinase 2 (ALK2), we found that GS domain phosphorylation, FKBP12 dissociation, and disease mutations all destabilize a D354-R375 salt-bridge, which normally acts as an electrostatic lock to prevent coordination of adenosine triphosphate (ATP) to the catalytic site. We developed a WAFEX-guided principal analysis and unraveled how phosphorylation destabilizes this highly conserved salt-bridge in temporal and physical space. Using current-flow betweenness scores, we identified an allosteric network of residue-residue contacts between the GS domain and the catalytic site that controls the formation and disruption of this salt bridge. Importantly, our novel network analysis approach revealed how certain disease-causing mutations bypass FKBP12-mediated kinase inhibition to produce leaky signaling. We further provide experimental evidence that this salt-bridge lock exists in other STKR1s, and acts as a general safety mechanism in STKR1 to prevent pathological leaky signaling. In summary, our study provides a compelling and unifying allosteric activation mechanism in STKR1 kinases that reconciles a large number of experimental studies and sheds light on a novel therapeutic avenue to target disease-related STKR1 mutants.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Miyazono K., ten Dijke P., Yamashita H. & Heldin C. H. Signal transduction via serine/threonine kinase receptors. Semin Cell Biol 5, 389–398 (1994). - PubMed
-
- Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol 13, 616–630, doi: 10.1038/nrm3434 (2012). - DOI - PMC - PubMed
-
- Nohe A. et al. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem 277, 5330–5338, doi: 10.1074/jbc.M102750200 (2002). - DOI - PubMed
-
- Shi Y. & Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113, 685–700 (2003). - PubMed
-
- Mathews L. S. & Vale W. W. Characterization of type II activin receptors. Binding, processing, and phosphorylation. J Biol Chem 268, 19013–19018 (1993). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
