

Composite likelihood method for inferring local pedigrees
- PMID: 28827797
- PMCID: PMC5578687
- DOI: 10.1371/journal.pgen.1006963
Composite likelihood method for inferring local pedigrees
Abstract
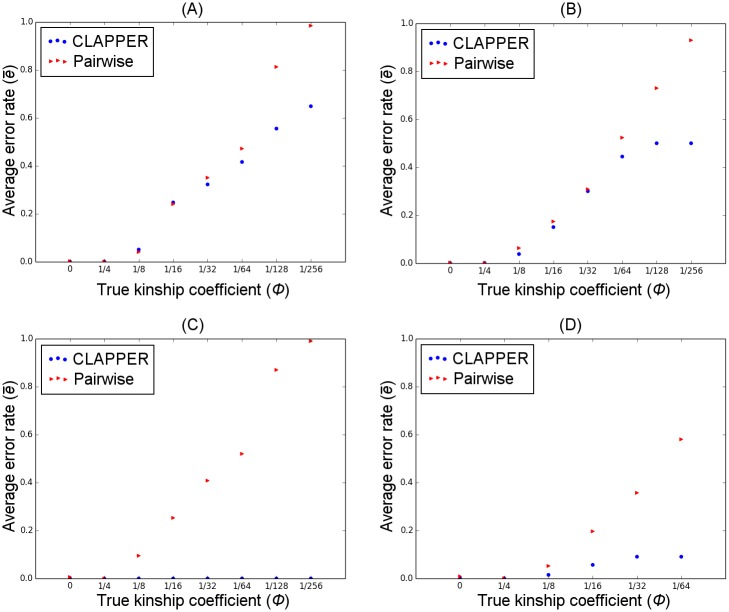
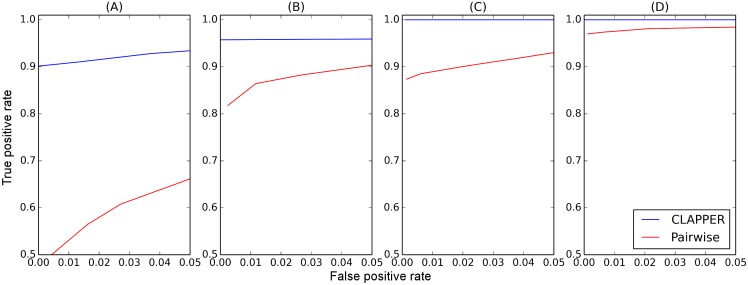
Pedigrees contain information about the genealogical relationships among individuals and are of fundamental importance in many areas of genetic studies. However, pedigrees are often unknown and must be inferred from genetic data. Despite the importance of pedigree inference, existing methods are limited to inferring only close relationships or analyzing a small number of individuals or loci. We present a simulated annealing method for estimating pedigrees in large samples of otherwise seemingly unrelated individuals using genome-wide SNP data. The method supports complex pedigree structures such as polygamous families, multi-generational families, and pedigrees in which many of the member individuals are missing. Computational speed is greatly enhanced by the use of a composite likelihood function which approximates the full likelihood. We validate our method on simulated data and show that it can infer distant relatives more accurately than existing methods. Furthermore, we illustrate the utility of the method on a sample of Greenlandic Inuit.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
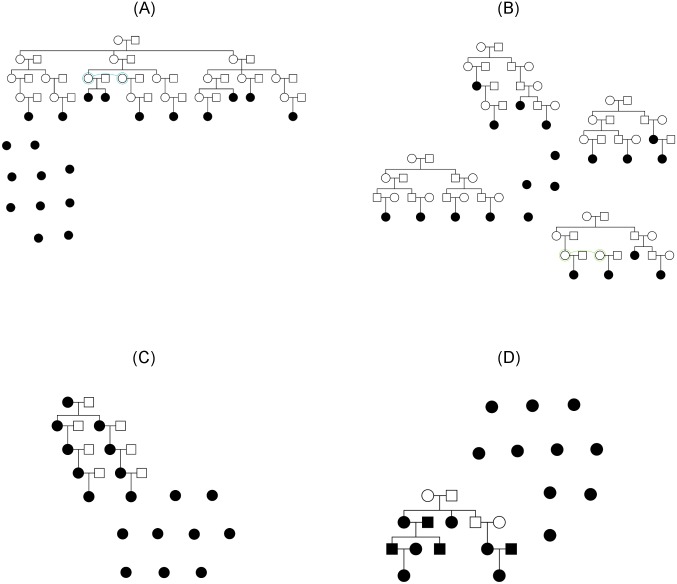
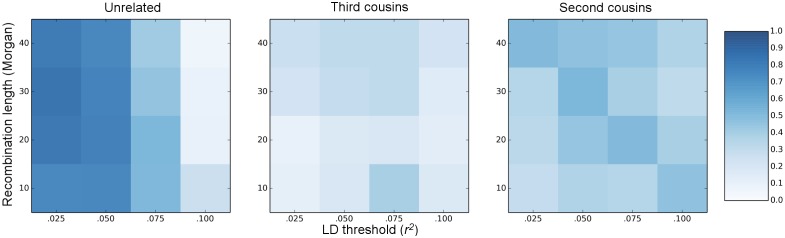
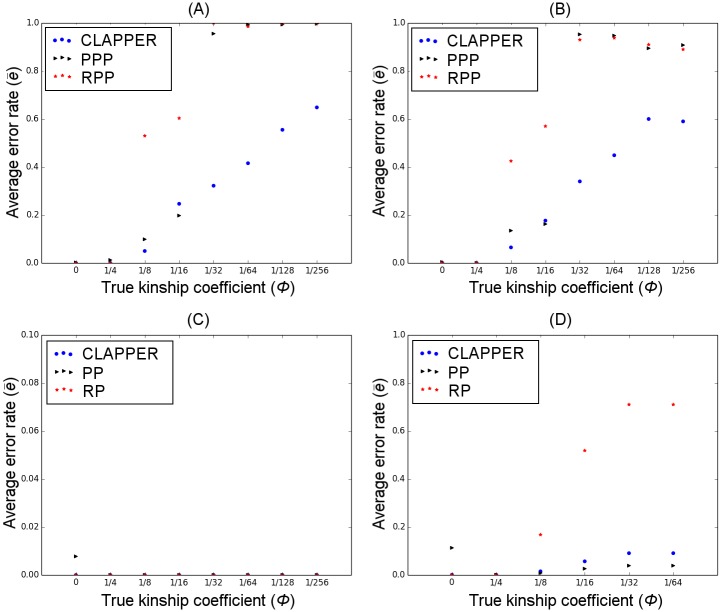
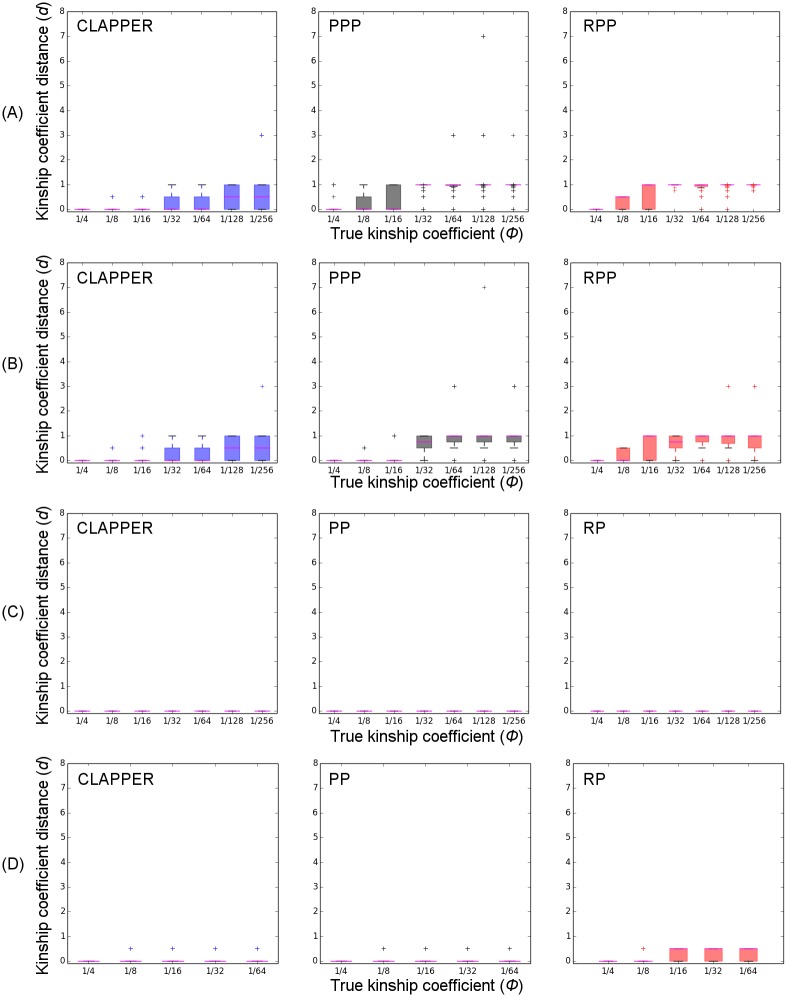
References
-
- Blouin MS. DNA-based methods for pedigree reconstruction and kinship analysis in natural populations. Trends Ecol Evol. 2003;18(10):503–511. 10.1016/S0169-5347(03)00225-8 - DOI
-
- Kingman JFC. The coalescent. Stochastic processes and their applications. 1982;13(3):235–248. 10.1016/0304-4149(82)90011-4 - DOI
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
