

Intracellular Proteolysis of Progranulin Generates Stable, Lysosomal Granulins that Are Haploinsufficient in Patients with Frontotemporal Dementia Caused by GRN Mutations
- PMID: 28828399
- PMCID: PMC5562298
- DOI: 10.1523/ENEURO.0100-17.2017
Intracellular Proteolysis of Progranulin Generates Stable, Lysosomal Granulins that Are Haploinsufficient in Patients with Frontotemporal Dementia Caused by GRN Mutations
Abstract
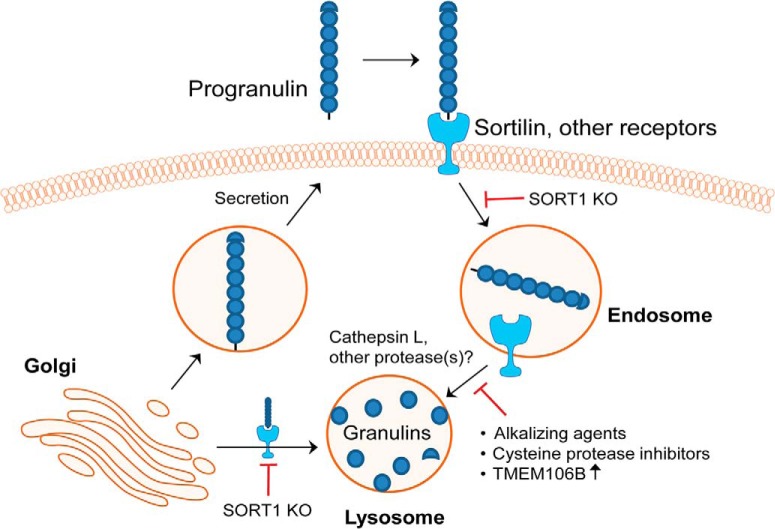
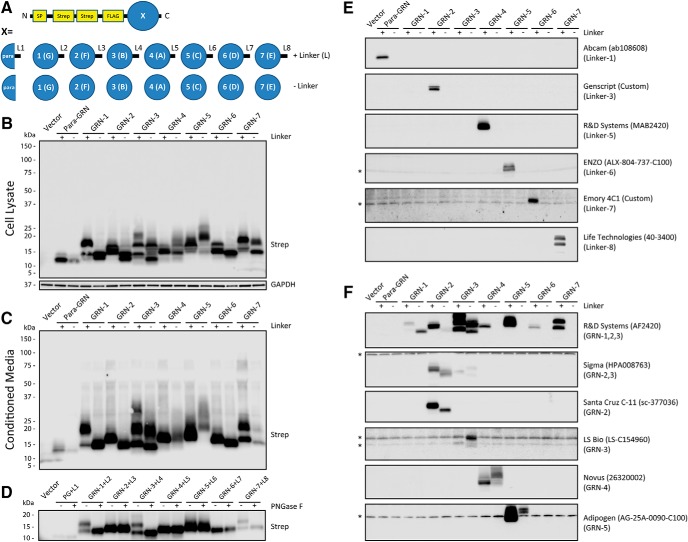
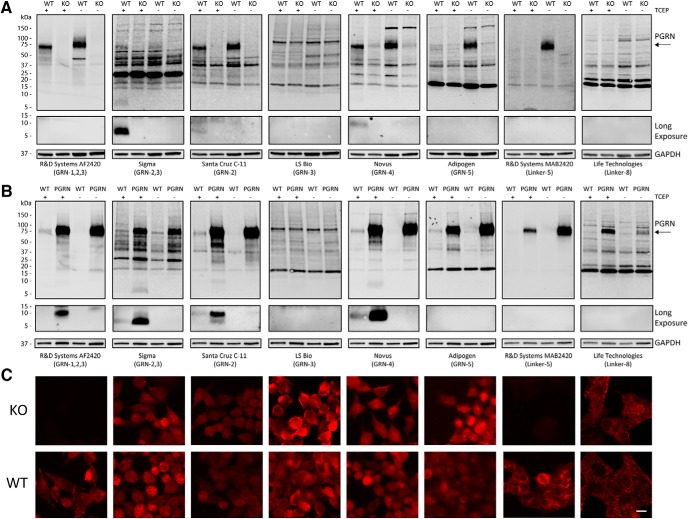
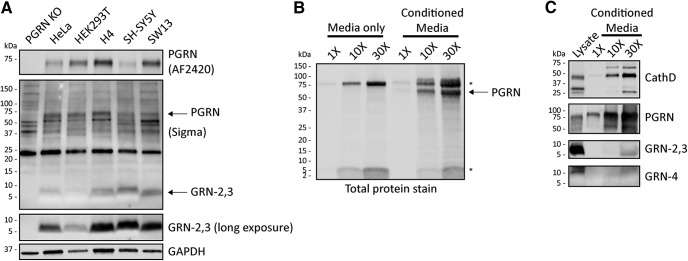
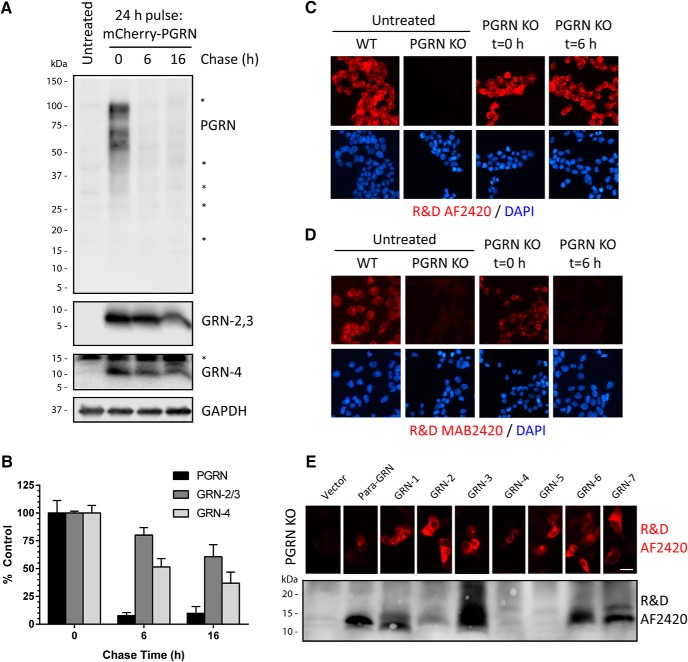
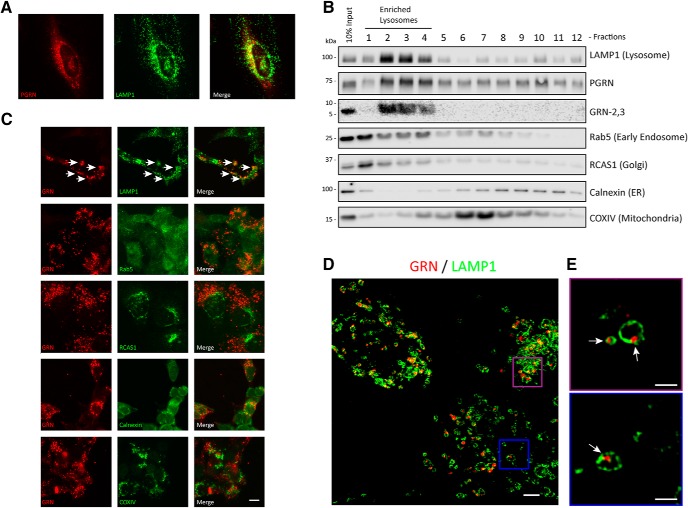
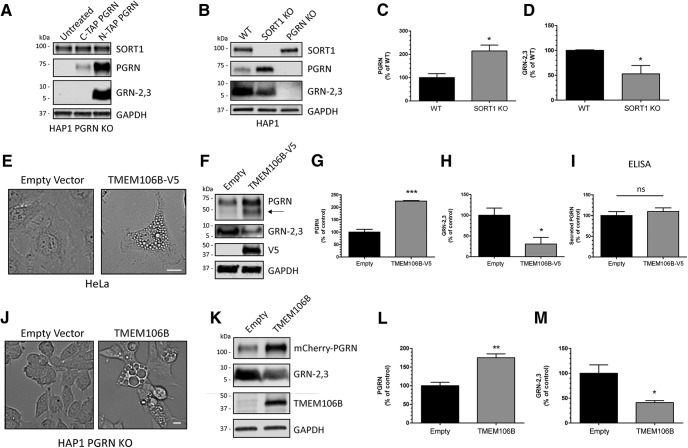
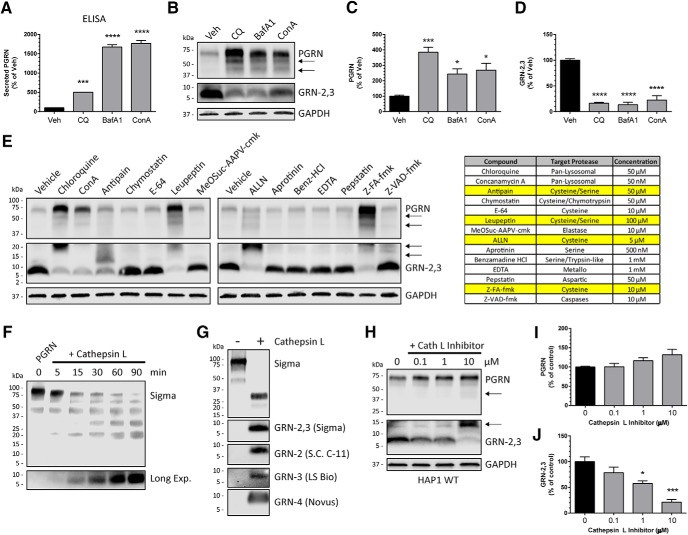
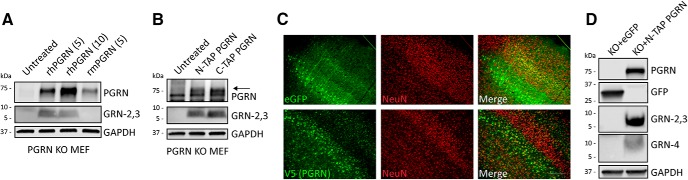
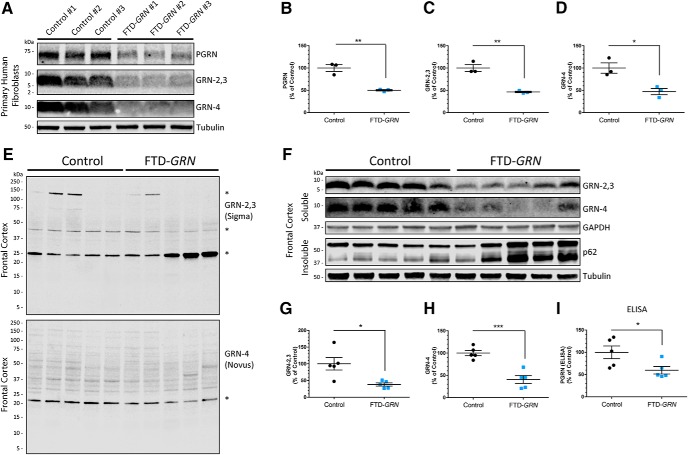
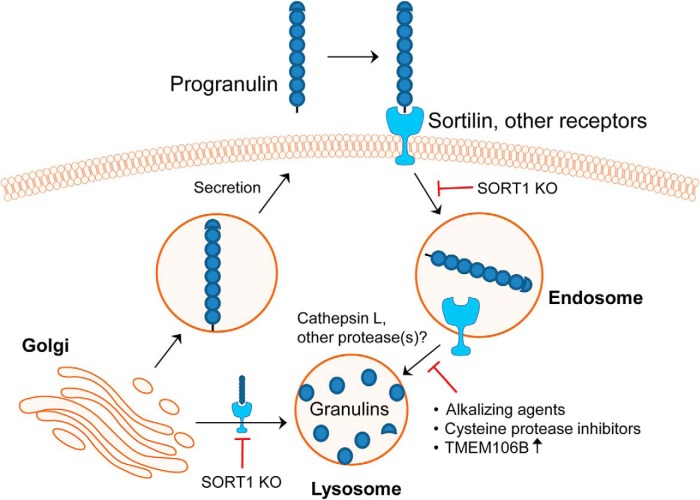
Homozygous or heterozygous mutations in the GRN gene, encoding progranulin (PGRN), cause neuronal ceroid lipofuscinosis (NCL) or frontotemporal dementia (FTD), respectively. NCL and FTD are characterized by lysosome dysfunction and neurodegeneration, indicating PGRN is important for lysosome homeostasis in the brain. PGRN is trafficked to the lysosome where its functional role is unknown. PGRN can be cleaved into seven 6-kDa proteins called granulins (GRNs); however, little is known about how GRNs are produced or if levels of GRNs are altered in FTD-GRN mutation carriers. Here, we report the identification and characterization of antibodies that reliably detect several human GRNs by immunoblot and immunocytochemistry. Using these tools, we find that endogenous GRNs are present within multiple cell lines and are constitutively produced. Further, extracellular PGRN is endocytosed and rapidly processed into stable GRNs within lysosomes. Processing of PGRN into GRNs is conserved between humans and mice and is modulated by sortilin expression and mediated by cysteine proteases (i.e. cathpesin L). Induced lysosome dysfunction caused by alkalizing agents or increased expression of transmembrane protein 106B (TMEM106B) inhibit processing of PGRN into GRNs. Finally, we find that multiple GRNs are haploinsufficient in primary fibroblasts and cortical brain tissue from FTD-GRN patients. Taken together, our findings raise the interesting possibility that GRNs carry out critical lysosomal functions and that loss of GRNs should be explored as an initiating factor in lysosomal dysfunction and neurodegeneration caused by GRN mutations.
Keywords: Alzheimer's disease; Parkinson's disease; amyotophic lateral sclerosis; autophagy; cathepsin L; frontotemporal dementia; granulins; lysosomal storage disease; neurodegeneration; neuroinflammation; neuronal ceroid lipofuscinosis; progranulin.
Figures
References
-
- Ahmed Z, Sheng H, Xu Y-f, Lin W-L, Innes AE, Gass J, Yu X, Hou H, Chiba S, Yamanouchi K, Leissring M, Petrucelli L, Nishihara M, Hutton ML, McGowan E, Dickson DW, Lewis J (2010) Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol 177:311–324. 10.2353/ajpath.2010.090915 - DOI - PMC - PubMed
-
- Almeida MR, Macário MC, Ramos L, Baldeiras I, Ribeiro MH, Santana I (2016) Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging 41:200.e1–200.e5. 10.1016/j.neurobiolaging.2016.02.019 - DOI - PubMed
-
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, et al. (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919. 10.1038/nature05016 - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous