

High-density lipoproteins suppress Aβ-induced PBMC adhesion to human endothelial cells in bioengineered vessels and in monoculture
- PMID: 28830501
- PMCID: PMC5568306
- DOI: 10.1186/s13024-017-0201-0
High-density lipoproteins suppress Aβ-induced PBMC adhesion to human endothelial cells in bioengineered vessels and in monoculture
Abstract
Background: Alzheimer's Disease (AD), characterized by accumulation of beta-amyloid (Aβ) plaques in the brain, can be caused by age-related failures to clear Aβ from the brain through pathways that involve the cerebrovasculature. Vascular risk factors are known to increase AD risk, but less is known about potential protective factors. We hypothesize that high-density lipoproteins (HDL) may protect against AD, as HDL have vasoprotective properties that are well described for peripheral vessels. Epidemiological studies suggest that HDL is associated with reduced AD risk, and animal model studies support a beneficial role for HDL in selectively reducing cerebrovascular amyloid deposition and neuroinflammation. However, the mechanism by which HDL may protect the cerebrovascular endothelium in the context of AD is not understood.
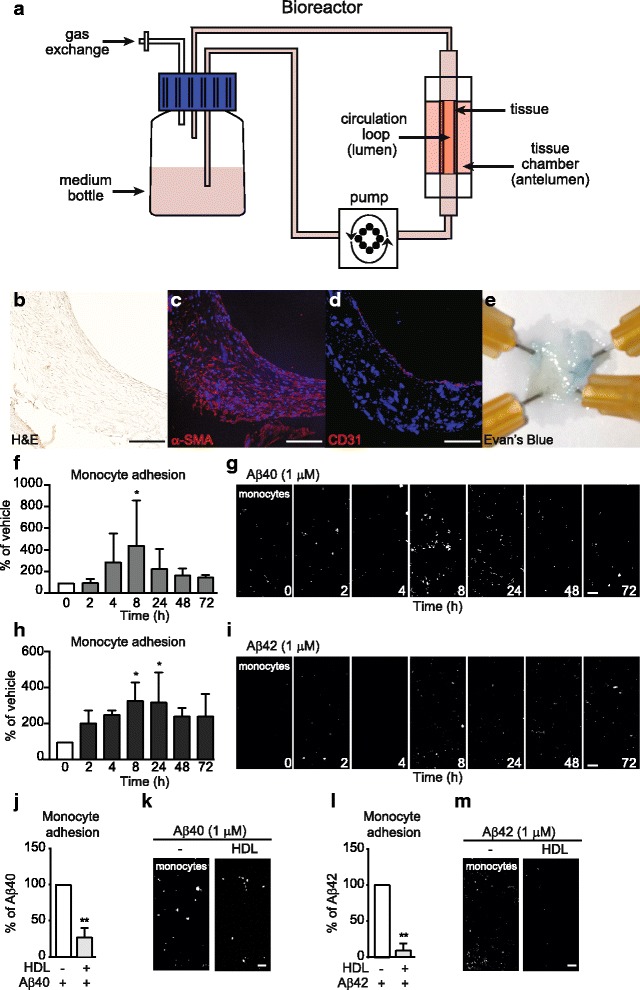
Methods: We used peripheral blood mononuclear cell adhesion assays in both a highly novel three dimensional (3D) biomimetic model of the human vasculature composed of primary human endothelial cells (EC) and smooth muscle cells cultured under flow conditions, as well as in monolayer cultures of ECs, to study how HDL protects ECs from the detrimental effects of Aβ.
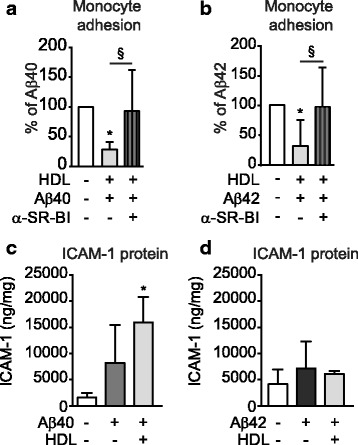
Results: Following Aβ addition to the abluminal (brain) side of the vessel, we demonstrate that HDL circulated within the lumen attenuates monocyte adhesion to ECs in this biofidelic vascular model. The mechanism by which HDL suppresses Aβ-mediated monocyte adhesion to ECs was investigated using monotypic EC cultures. We show that HDL reduces Aβ-induced PBMC adhesion to ECs independent of nitric oxide (NO) production, miR-233 and changes in adhesion molecule expression. Rather, HDL acts through scavenger receptor (SR)-BI to block Aβ uptake into ECs and, in cell-free assays, can maintain Aβ in a soluble state. We confirm the role of SR-BI in our bioengineered human vessel.
Conclusion: Our results define a novel activity of HDL that suppresses Aβ-mediated monocyte adhesion to the cerebrovascular endothelium.
Keywords: Alzheimer’s disease; Beta-amyloid; Endothelial cells; Engineered vessel; High-density lipoprotein.
Conflict of interest statement
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures





References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
