

Adult-Onset Vitelliform Macular Dystrophy caused by BEST1 p.Ile38Ser Mutation is a Mild Form of Best Vitelliform Macular Dystrophy
- PMID: 28831140
- PMCID: PMC5567297
- DOI: 10.1038/s41598-017-09629-9
Adult-Onset Vitelliform Macular Dystrophy caused by BEST1 p.Ile38Ser Mutation is a Mild Form of Best Vitelliform Macular Dystrophy
Abstract
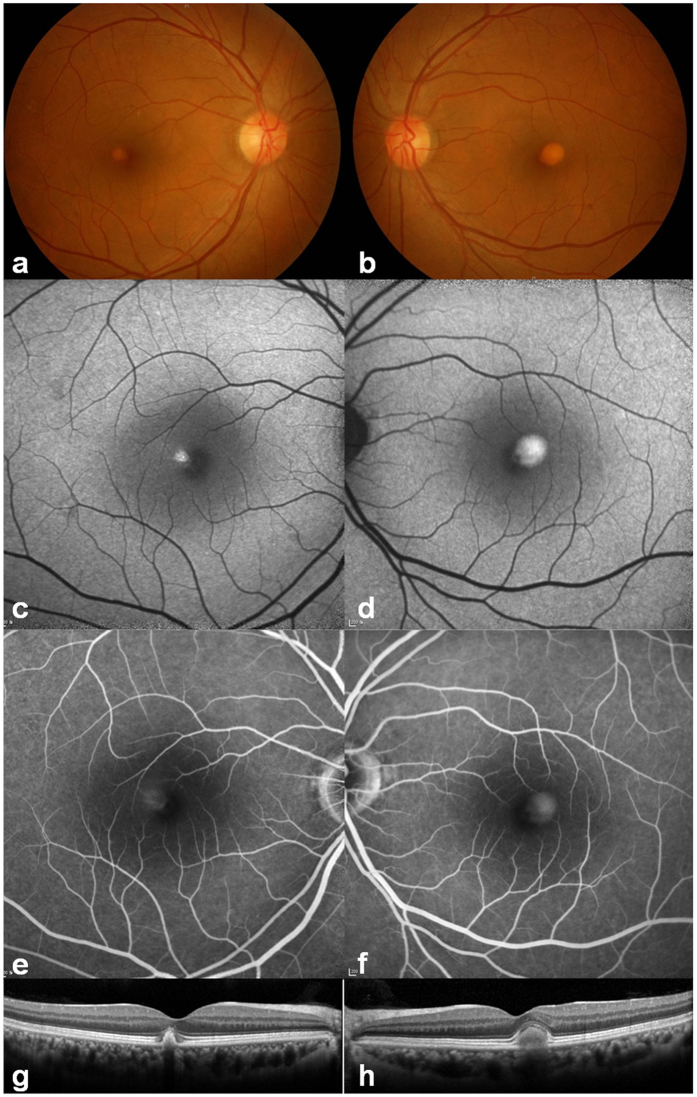
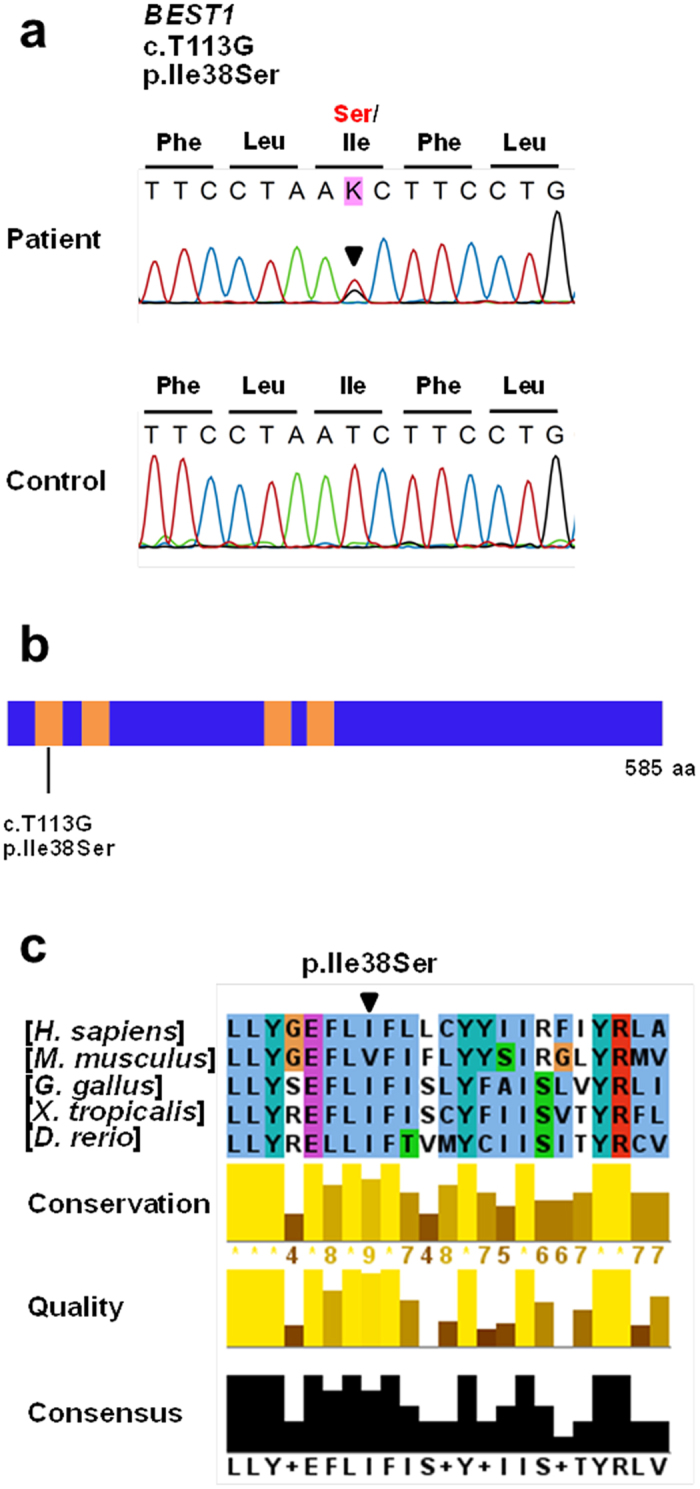
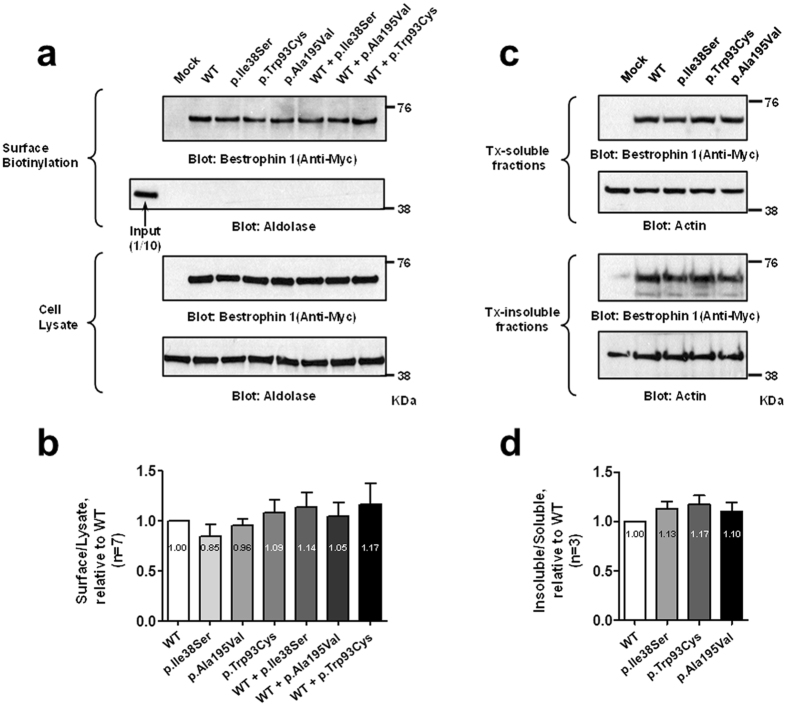
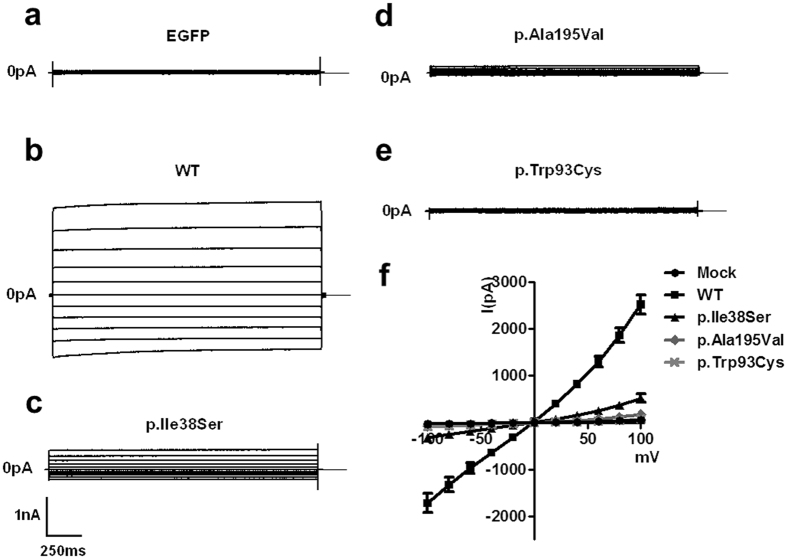
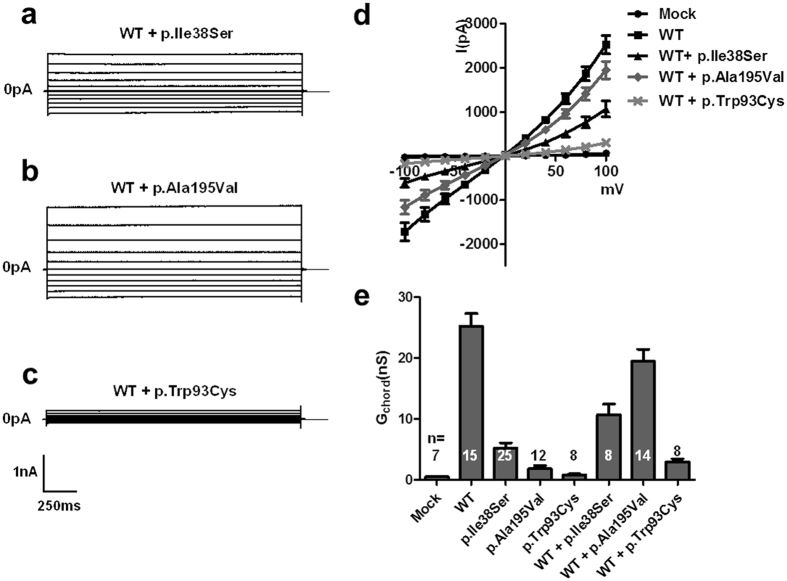
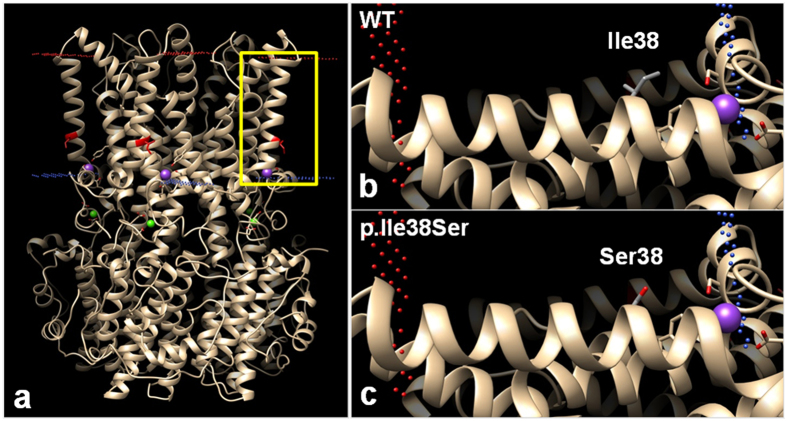
Adult-onset vitelliform macular dystrophy (AVMD) is a common and benign macular degeneration which can be caused by BEST1 mutation. Here, we investigated the clinical characteristics associated with a newly identified BEST1 mutation, p.Ile38Ser and confirmed the associated physiological functional defects. The 51-year-old patient presented bilateral small subretinal yellow deposits. Consistent with AVMD, the corresponding lesions showed hyperautofluorescence, late staining in fluorescein angiography, and subretinal hyper-reflective materials in spectral-domain optical coherence tomography. Genetic analysis demonstrated that the patient presented with a heterozygous p.Ile38Ser BEST1 mutation. Surface biotinylation and patch clamp experiments were performed in transfected HEK293T cells. Although, the identified BEST1 mutant maintains normal membrane expression, p.Ile38Ser mutant showed significantly smaller currents than wild type (WT). However, it showed larger currents than other BEST1 mutants, p.Trp93Cys, causing autosomal dominant best vitelliform macular dystrophy (BVMD), and p.Ala195Val, causing autosomal recessive bestrophinopathy (ARB). The cells expressing both WT and each BEST1 mutant showed that the functional defect of p.Ile38ser was milder than that of p.Trp93Cys, whereas combination of p.Ala195Val with WT showed good current. We identified and described the phenotype and in vitro functions of a novel BEST1 mutation causing AVMD. AVMD induced by p.Ile38Ser BEST1 mutation is a mild form of BVMD.
Conflict of interest statement
E.K.K. is the medical advisory board member of the Avellino LAB USA. Other authors have no conflict of interest to declare.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
