

SweGen: a whole-genome data resource of genetic variability in a cross-section of the Swedish population
- PMID: 28832569
- PMCID: PMC5765326
- DOI: 10.1038/ejhg.2017.130
SweGen: a whole-genome data resource of genetic variability in a cross-section of the Swedish population
Abstract
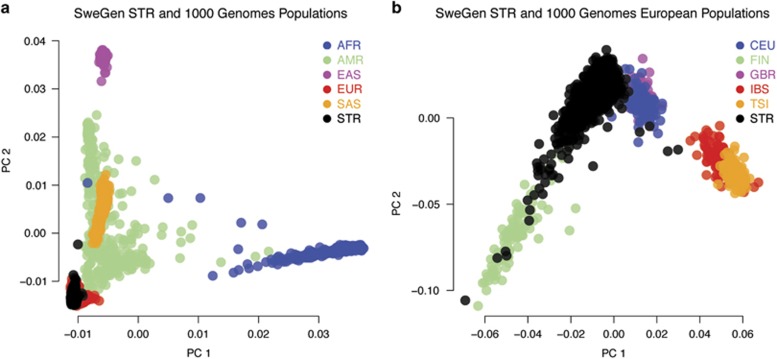
Here we describe the SweGen data set, a comprehensive map of genetic variation in the Swedish population. These data represent a basic resource for clinical genetics laboratories as well as for sequencing-based association studies by providing information on genetic variant frequencies in a cohort that is well matched to national patient cohorts. To select samples for this study, we first examined the genetic structure of the Swedish population using high-density SNP-array data from a nation-wide cohort of over 10 000 Swedish-born individuals included in the Swedish Twin Registry. A total of 1000 individuals, reflecting a cross-section of the population and capturing the main genetic structure, were selected for whole-genome sequencing. Analysis pipelines were developed for automated alignment, variant calling and quality control of the sequencing data. This resulted in a genome-wide collection of aggregated variant frequencies in the Swedish population that we have made available to the scientific community through the website https://swefreq.nbis.se. A total of 29.2 million single-nucleotide variants and 3.8 million indels were detected in the 1000 samples, with 9.9 million of these variants not present in current databases. Each sample contributed with an average of 7199 individual-specific variants. In addition, an average of 8645 larger structural variants (SVs) were detected per individual, and we demonstrate that the population frequencies of these SVs can be used for efficient filtering analyses. Finally, our results show that the genetic diversity within Sweden is substantial compared with the diversity among continental European populations, underscoring the relevance of establishing a local reference data set.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Gudbjartsson DF, Helgason H, Gudjonsson SA et al: Large-scale whole-genome sequencing of the Icelandic population. Nat Genet 2015; 47: 435–444. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
