

Hyperhomocysteinaemia and vascular injury: advances in mechanisms and drug targets
- PMID: 28836260
- PMCID: PMC5867019
- DOI: 10.1111/bph.13988
Hyperhomocysteinaemia and vascular injury: advances in mechanisms and drug targets
Abstract
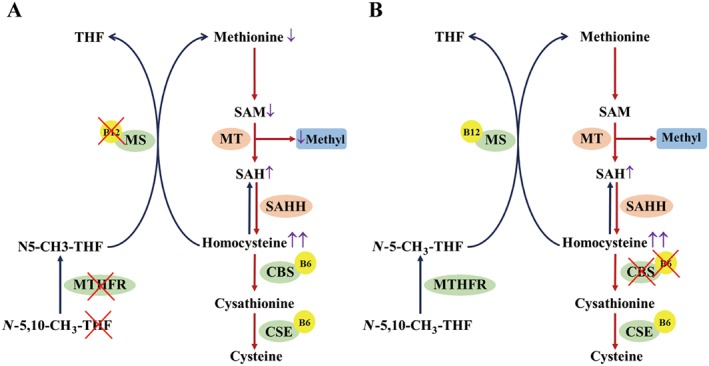
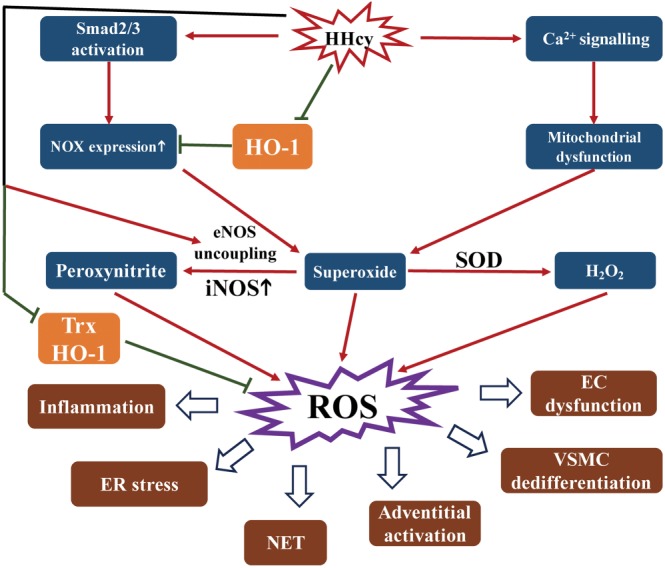
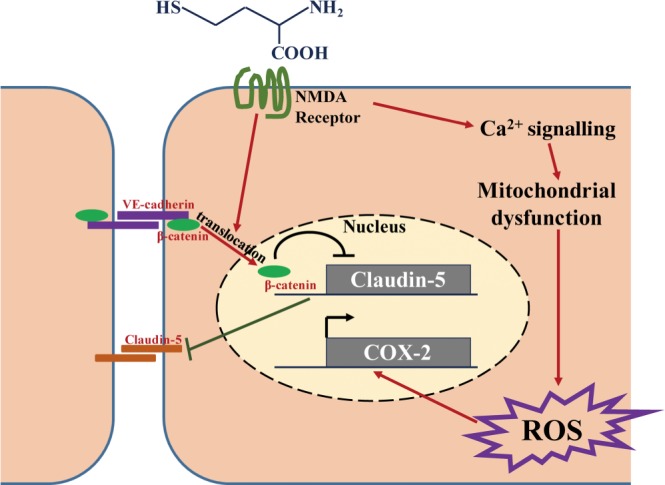
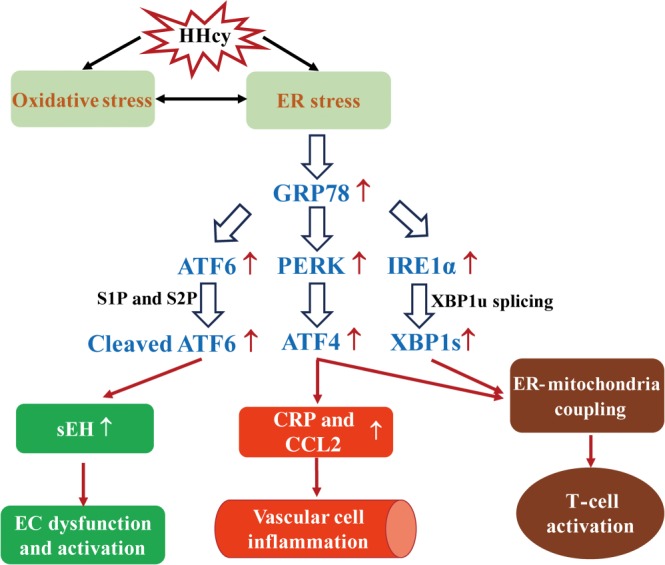
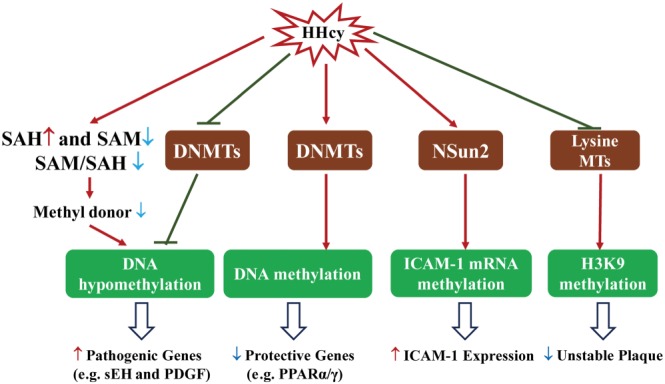
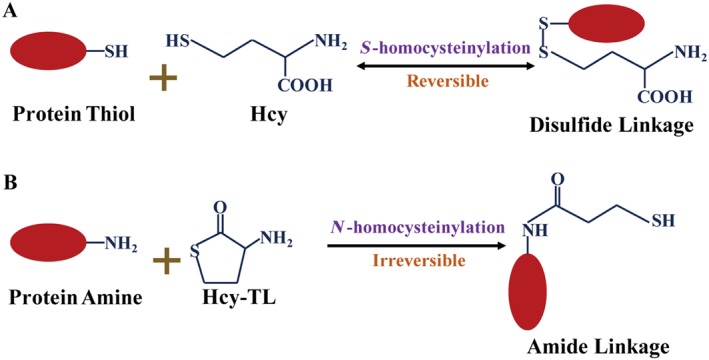
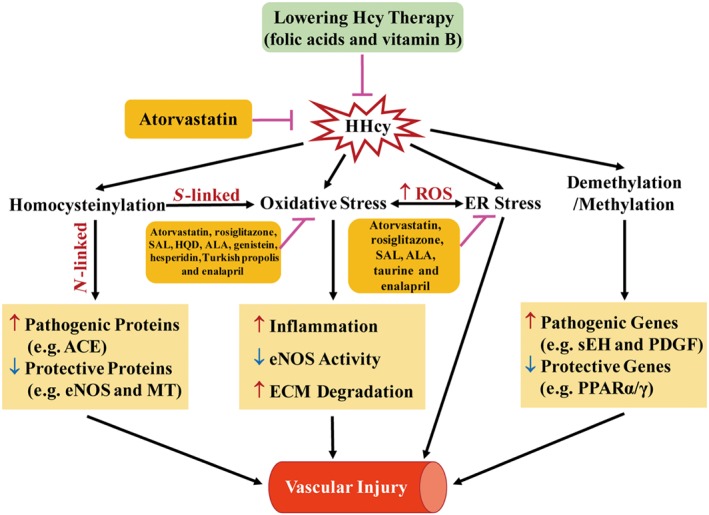
Homocysteine is a sulphur-containing non-proteinogenic amino acid. Hyperhomocysteinaemia (HHcy), the pathogenic elevation of plasma homocysteine as a result of an imbalance of its metabolism, is an independent risk factor for various vascular diseases, such as atherosclerosis, hypertension, vascular calcification and aneurysm. Treatments aimed at lowering plasma homocysteine via dietary supplementation with folic acids and vitamin B are more effective in preventing vascular disease where the population has a normally low folate consumption than in areas with higher dietary folate. To date, the mechanisms of HHcy-induced vascular injury are not fully understood. HHcy increases oxidative stress and its downstream signalling pathways, resulting in vascular inflammation. HHcy also causes vascular injury via endoplasmic reticulum stress. Moreover, HHcy up-regulates pathogenic genes and down-regulates protective genes via DNA demethylation and methylation respectively. Homocysteinylation of proteins induced by homocysteine also contributes to vascular injury by modulating intracellular redox state and altering protein function. Furthermore, HHcy-induced vascular injury leads to neuronal damage and disease. Also, an HHcy-activated sympathetic system and HHcy-injured adipose tissue also cause vascular injury, thus demonstrating the interactions between the organs injured by HHcy. Here, we have summarized the recent developments in the mechanisms of HHcy-induced vascular injury, which are further considered as potential therapeutic targets in this condition.
Linked articles: This article is part of a themed section on Spotlight on Small Molecules in Cardiovascular Diseases. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.8/issuetoc.
© 2017 The British Pharmacological Society.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
