

CD55 regulates self-renewal and cisplatin resistance in endometrioid tumors
- PMID: 28838952
- PMCID: PMC5584126
- DOI: 10.1084/jem.20170438
CD55 regulates self-renewal and cisplatin resistance in endometrioid tumors
Abstract
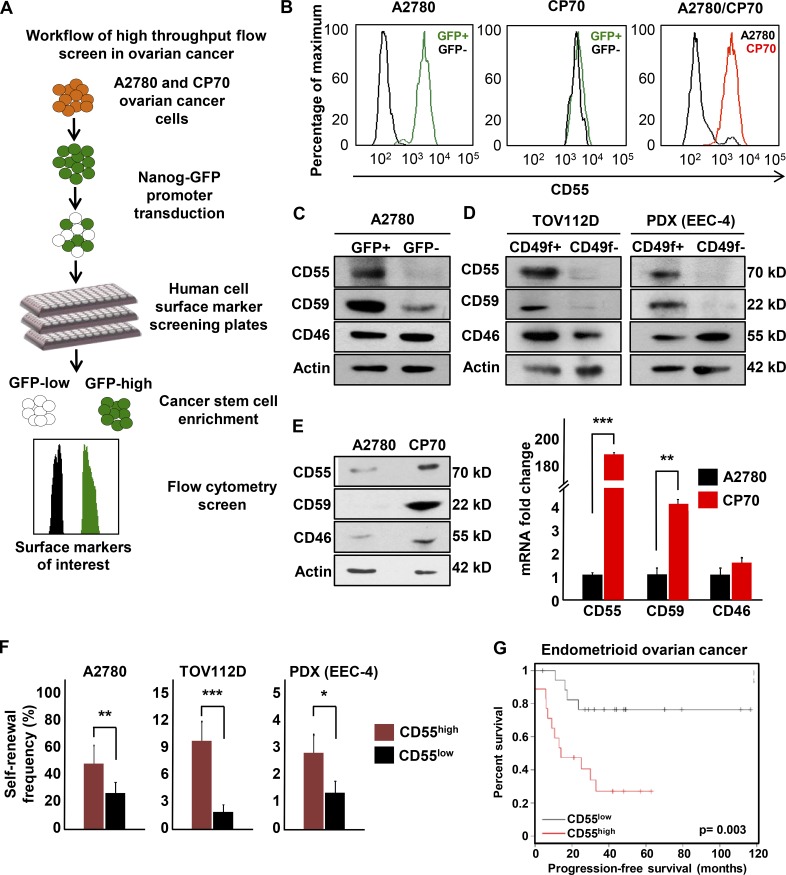
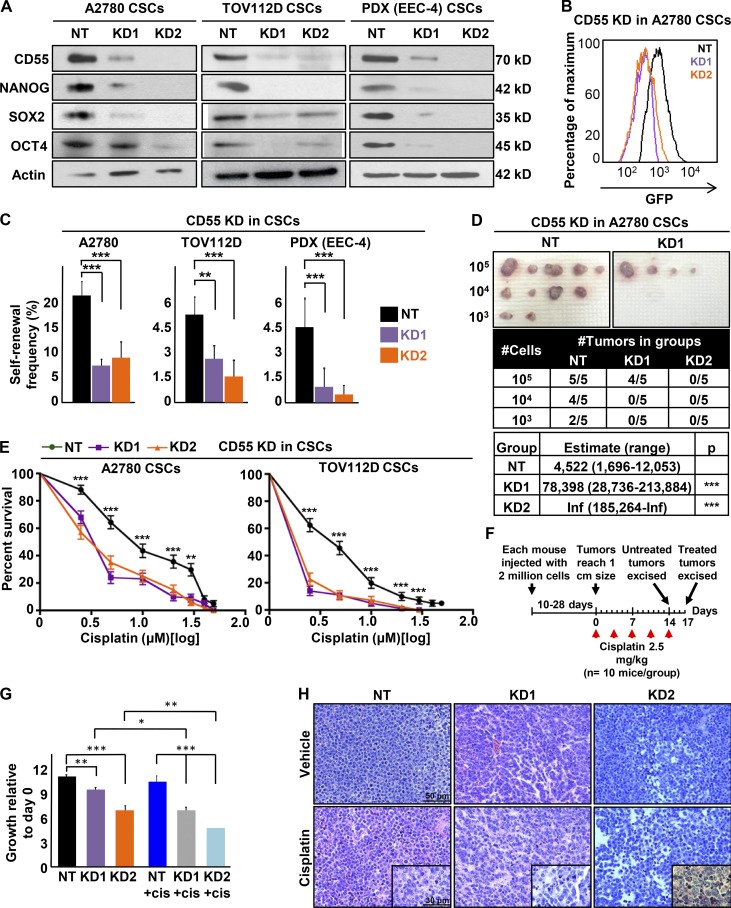
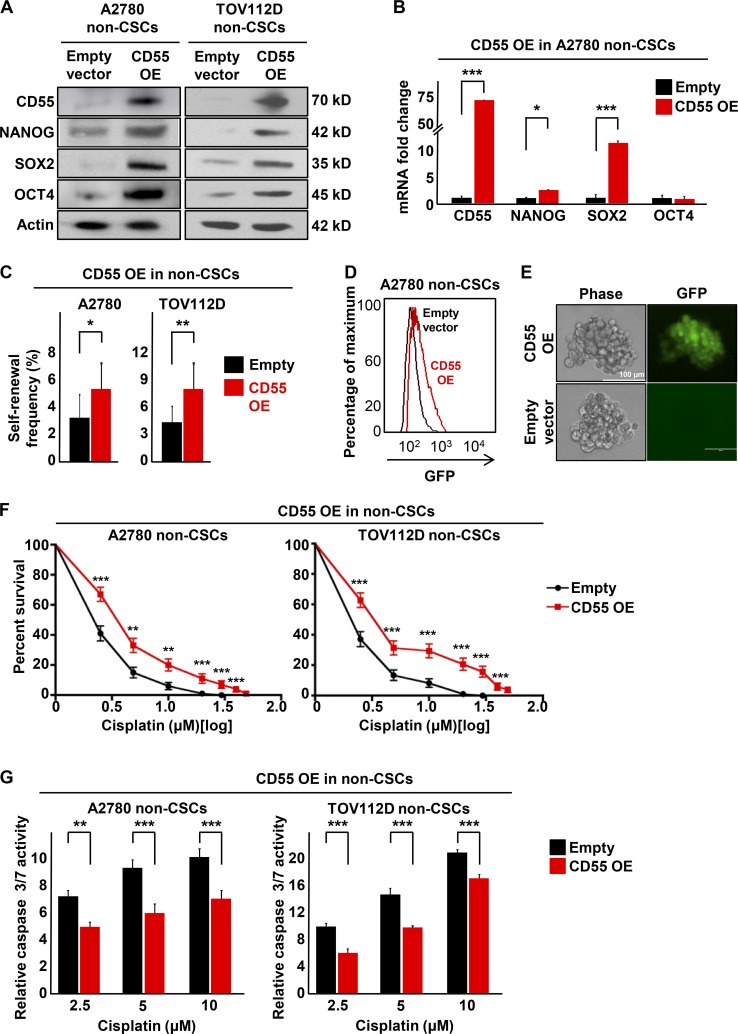
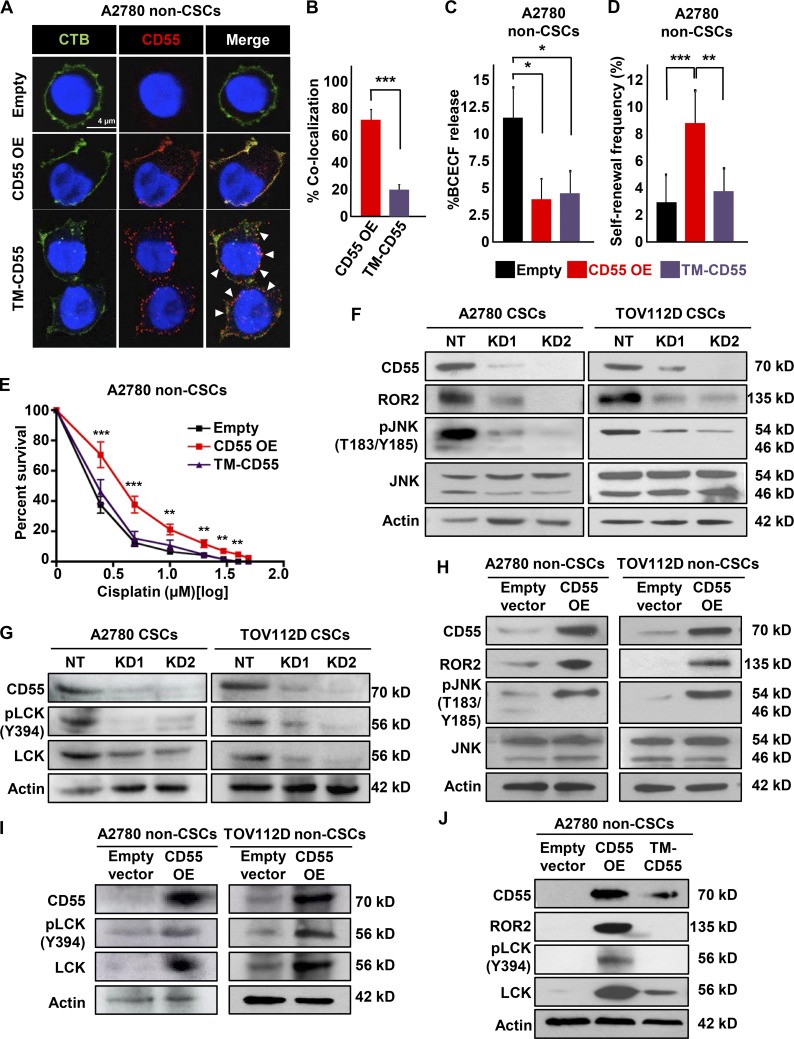
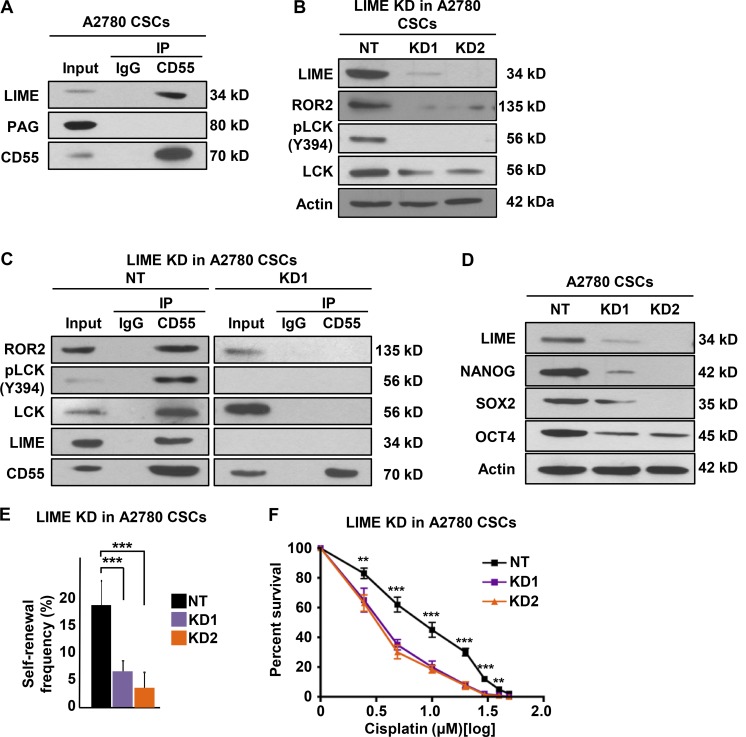
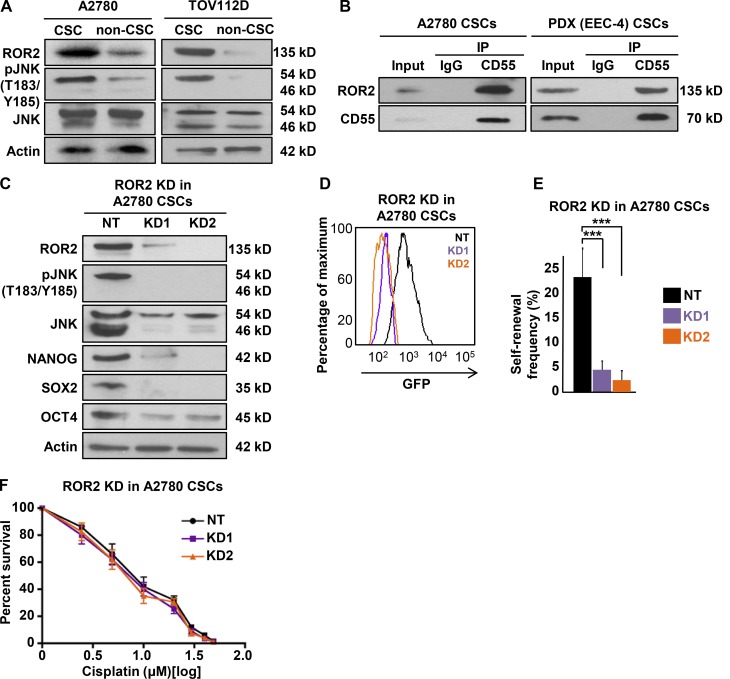
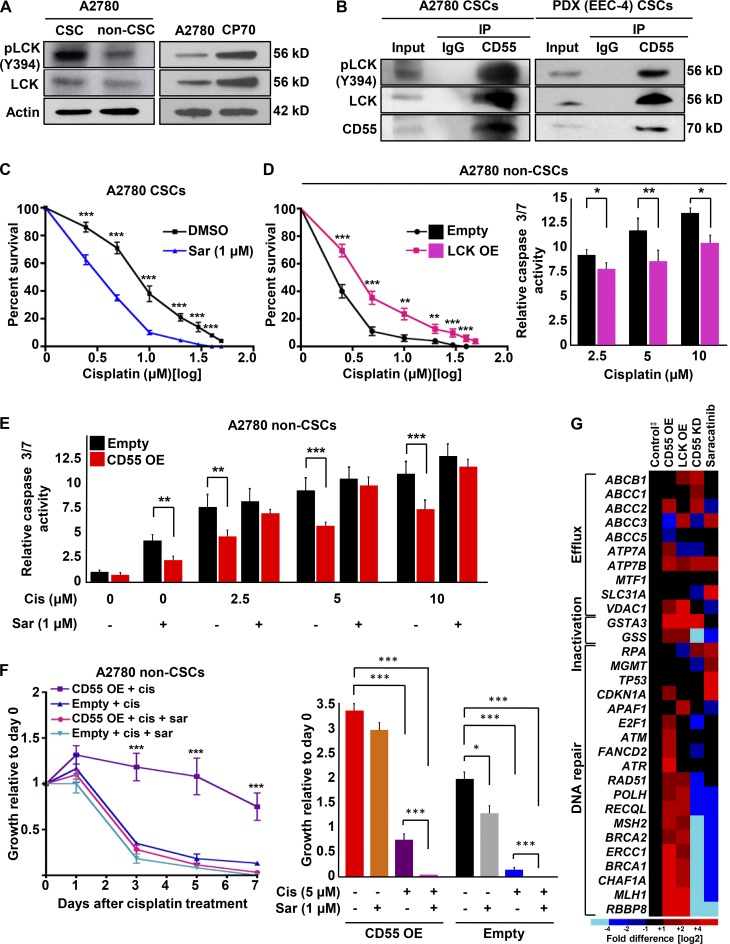
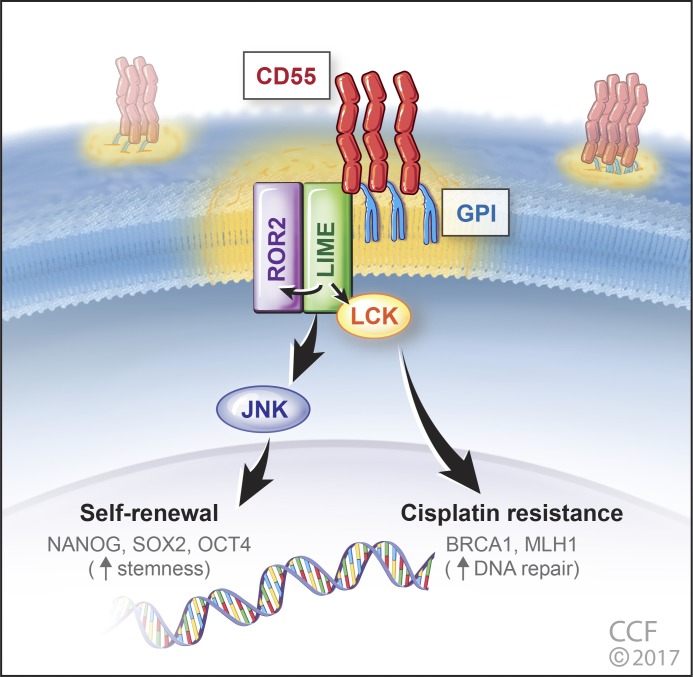
Effective targeting of cancer stem cells (CSCs) requires neutralization of self-renewal and chemoresistance, but these phenotypes are often regulated by distinct molecular mechanisms. Here we report the ability to target both of these phenotypes via CD55, an intrinsic cell surface complement inhibitor, which was identified in a comparative analysis between CSCs and non-CSCs in endometrioid cancer models. In this context, CD55 functions in a complement-independent manner and required lipid raft localization for CSC maintenance and cisplatin resistance. CD55 regulated self-renewal and core pluripotency genes via ROR2/JNK signaling and in parallel cisplatin resistance via lymphocyte-specific protein tyrosine kinase (LCK) signaling, which induced DNA repair genes. Targeting LCK signaling via saracatinib, an inhibitor currently undergoing clinical evaluation, sensitized chemoresistant cells to cisplatin. Collectively, our findings identify CD55 as a unique signaling node that drives self-renewal and therapeutic resistance through a bifurcating signaling axis and provides an opportunity to target both signaling pathways in endometrioid tumors.
© 2017 Saygin et al.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
