

Structural and Chemical Biology of Terpenoid Cyclases
- PMID: 28841019
- PMCID: PMC5599884
- DOI: 10.1021/acs.chemrev.7b00287
Structural and Chemical Biology of Terpenoid Cyclases
Erratum in
-
Correction to Structural and Chemical Biology of Terpenoid Cyclases.Chem Rev. 2018 Dec 26;118(24):11795. doi: 10.1021/acs.chemrev.8b00682. Epub 2018 Dec 12. Chem Rev. 2018. PMID: 30540443 Free PMC article. No abstract available.
Abstract
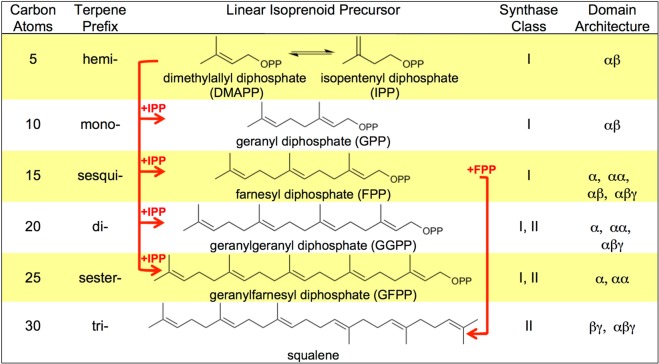
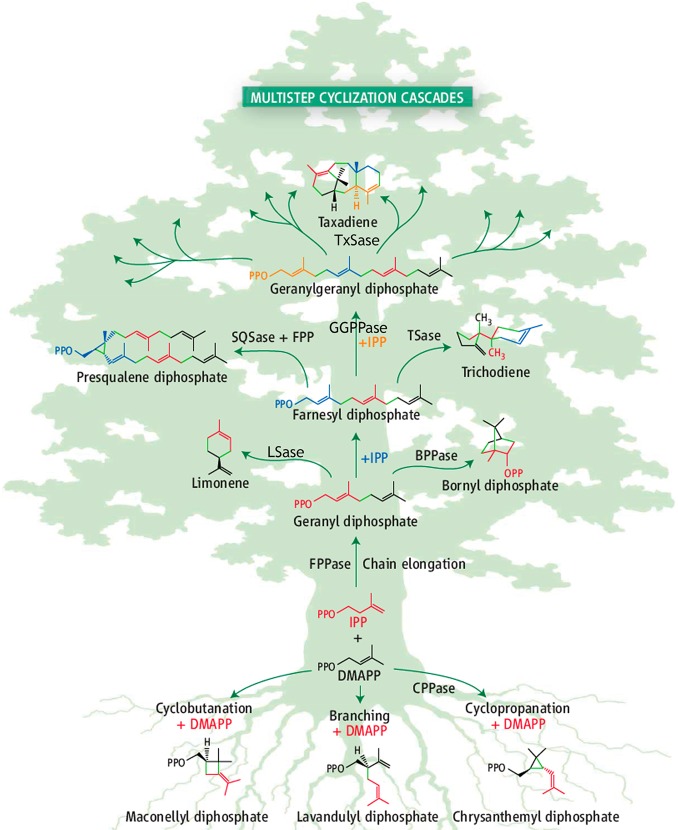
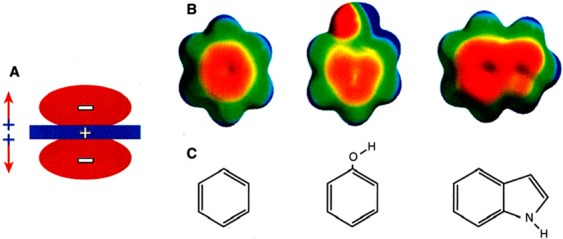
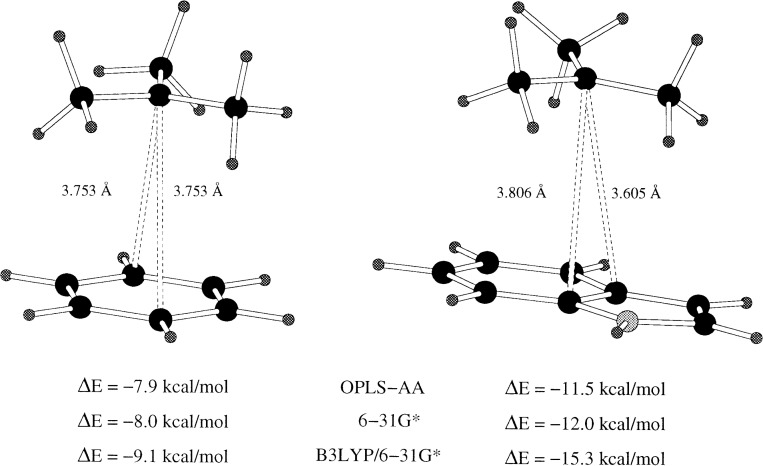
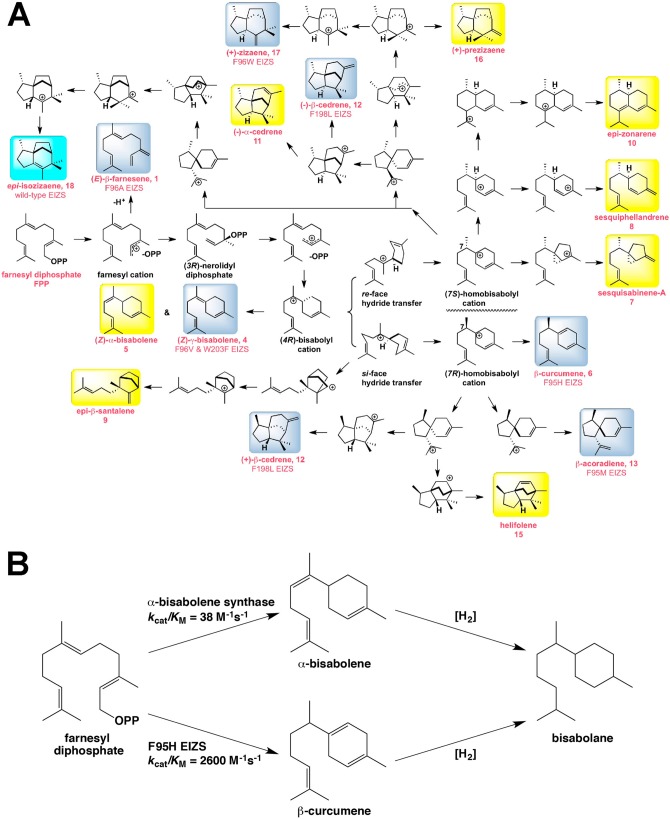
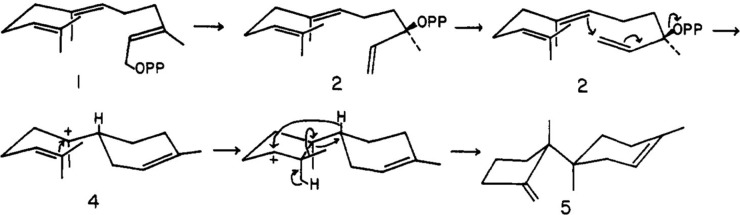
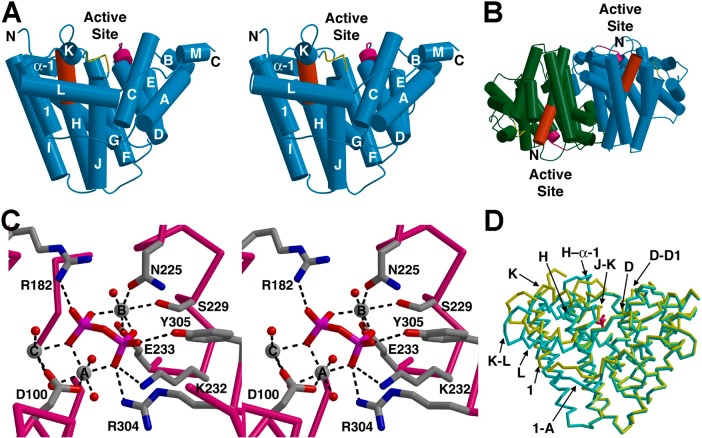
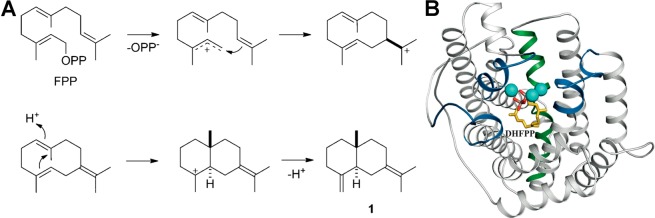
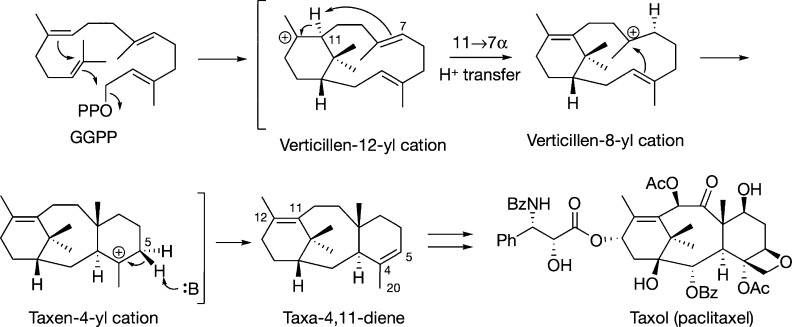
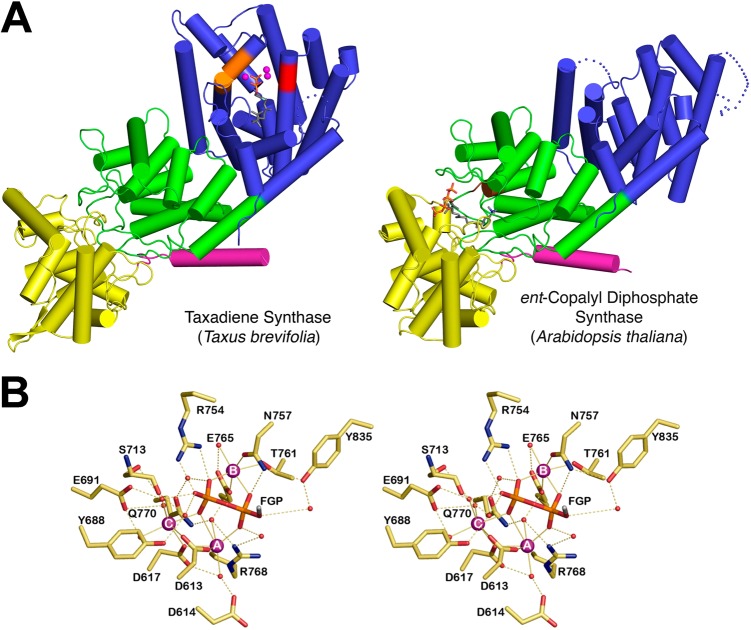
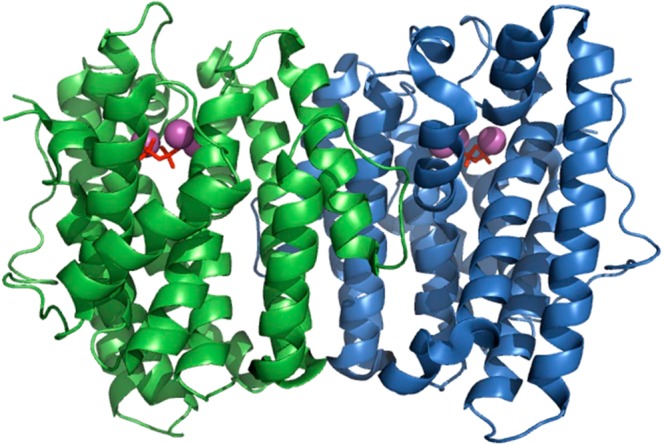
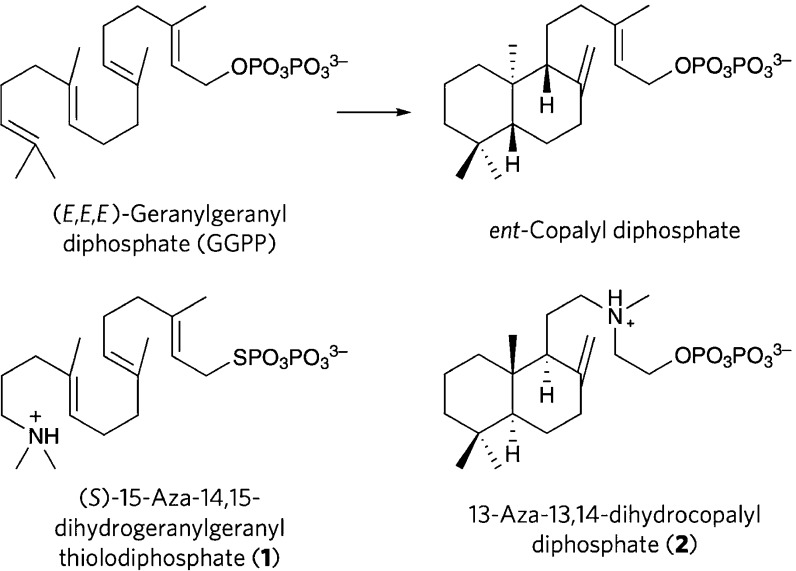
The year 2017 marks the twentieth anniversary of terpenoid cyclase structural biology: a trio of terpenoid cyclase structures reported together in 1997 were the first to set the foundation for understanding the enzymes largely responsible for the exquisite chemodiversity of more than 80000 terpenoid natural products. Terpenoid cyclases catalyze the most complex chemical reactions in biology, in that more than half of the substrate carbon atoms undergo changes in bonding and hybridization during a single enzyme-catalyzed cyclization reaction. The past two decades have witnessed structural, functional, and computational studies illuminating the modes of substrate activation that initiate the cyclization cascade, the management and manipulation of high-energy carbocation intermediates that propagate the cyclization cascade, and the chemical strategies that terminate the cyclization cascade. The role of the terpenoid cyclase as a template for catalysis is paramount to its function, and protein engineering can be used to reprogram the cyclization cascade to generate alternative and commercially important products. Here, I review key advances in terpenoid cyclase structural and chemical biology, focusing mainly on terpenoid cyclases and related prenyltransferases for which X-ray crystal structures have informed and advanced our understanding of enzyme structure and function.
Conflict of interest statement
The author declares no competing financial interest.
Figures
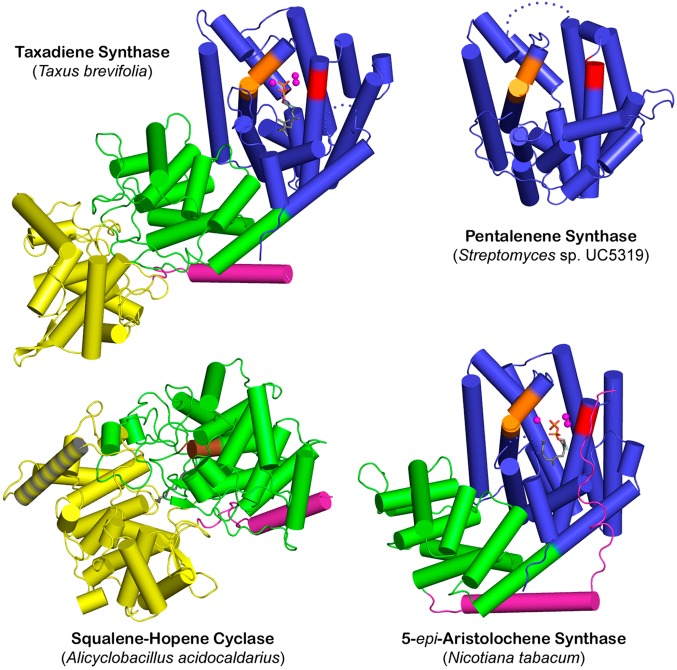
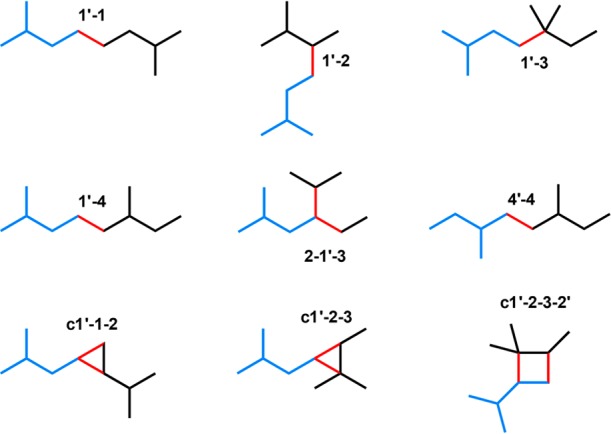
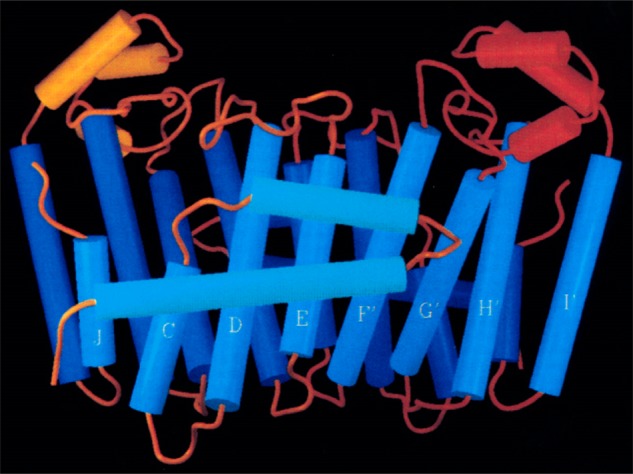
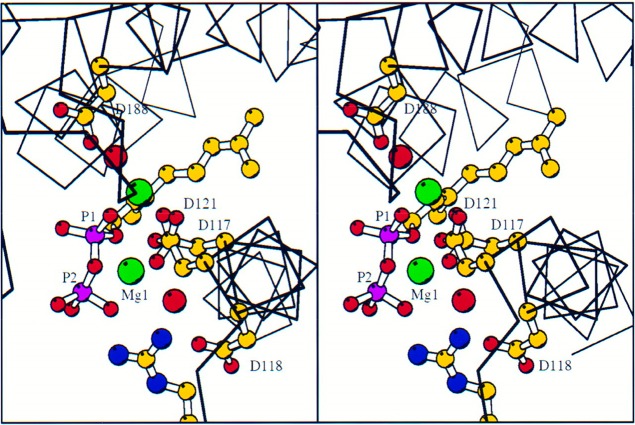
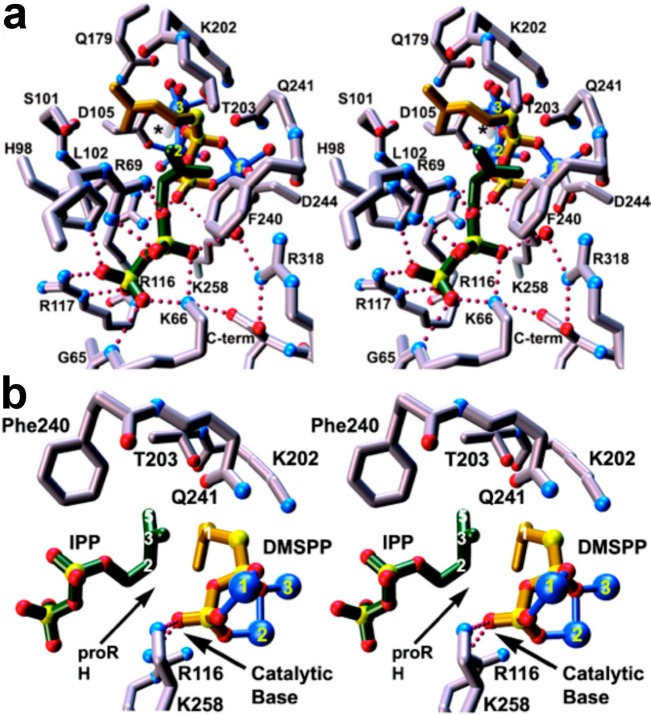
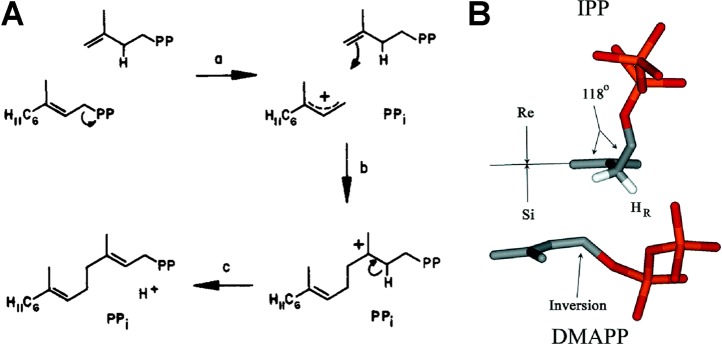
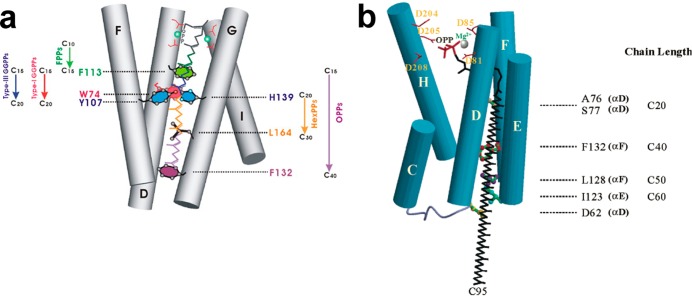
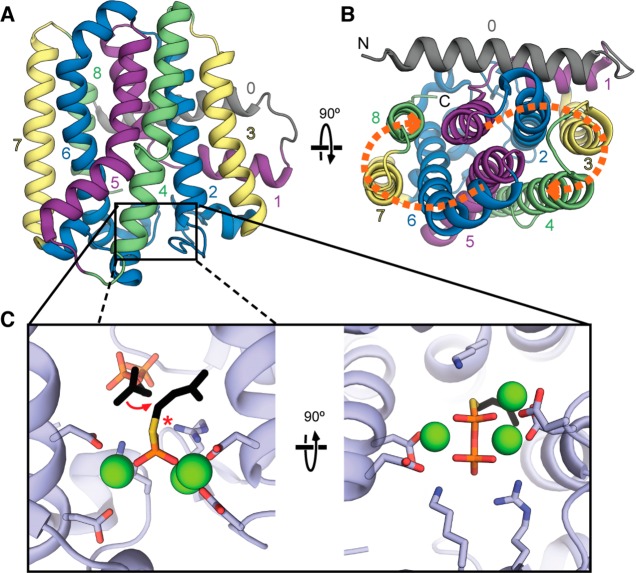
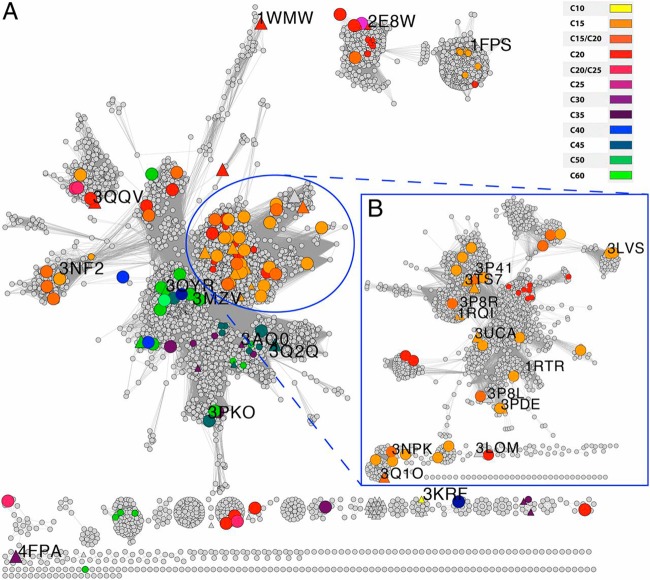
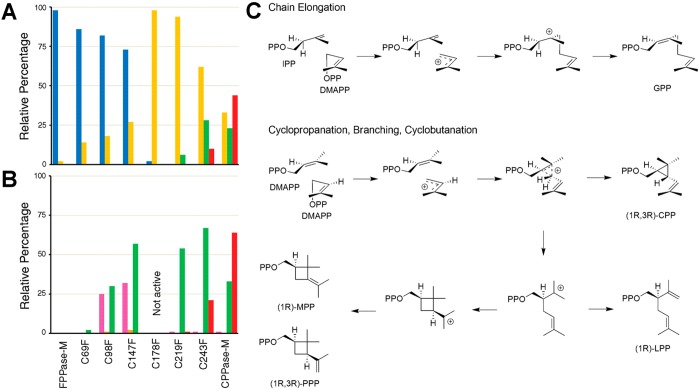
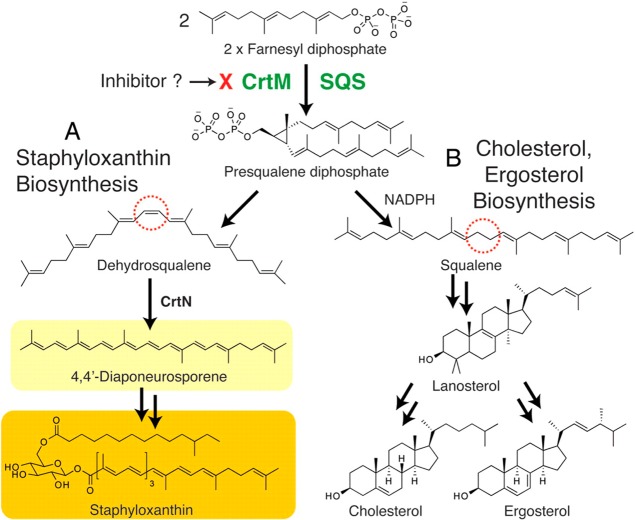
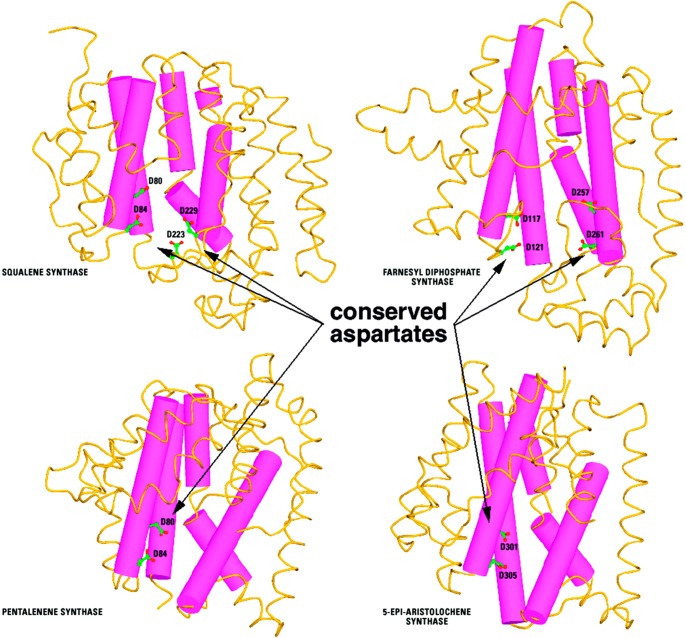
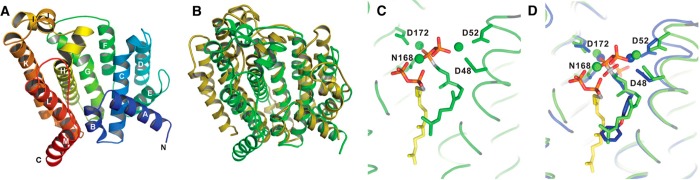
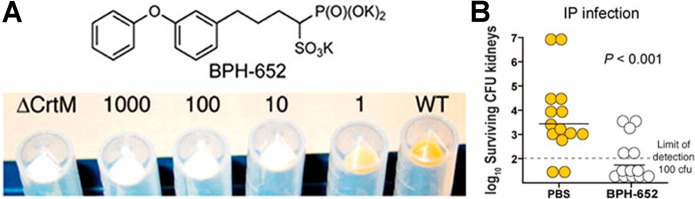
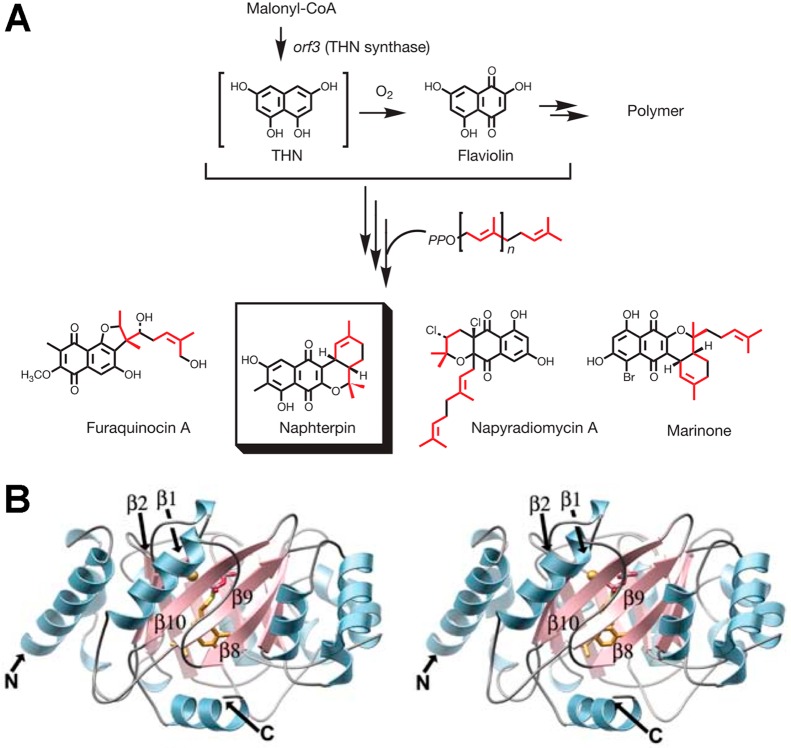
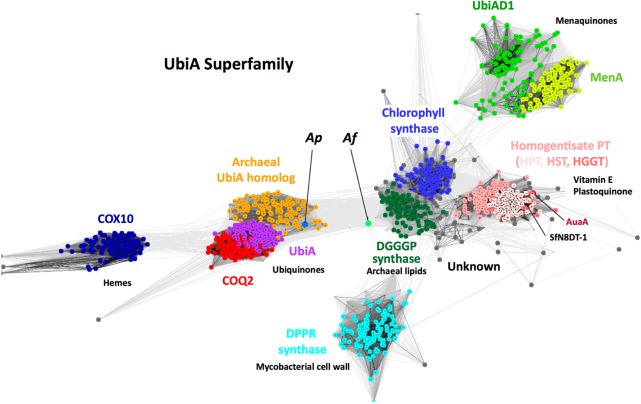
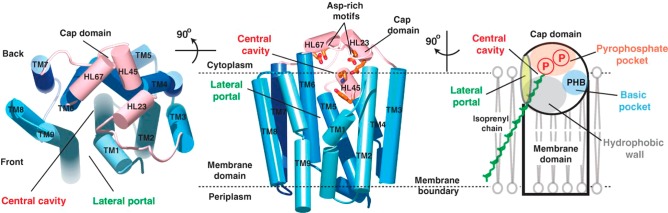
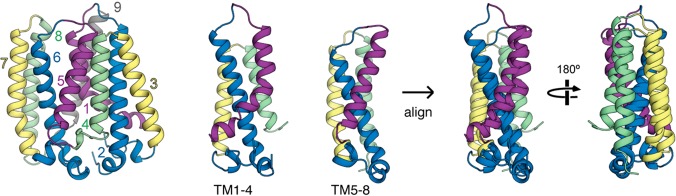
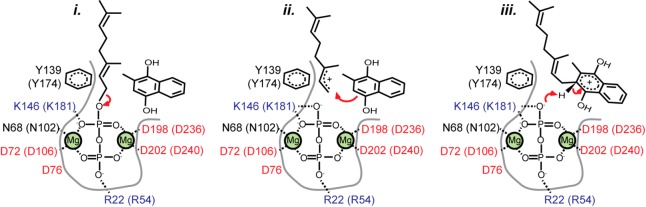
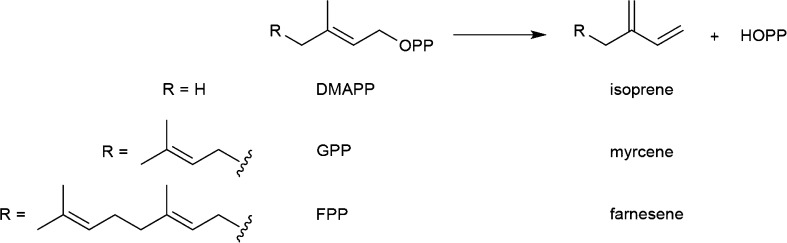

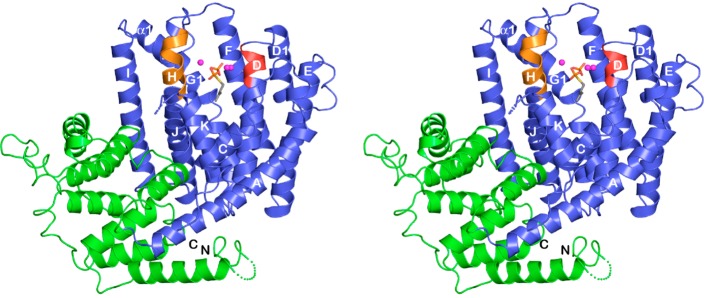
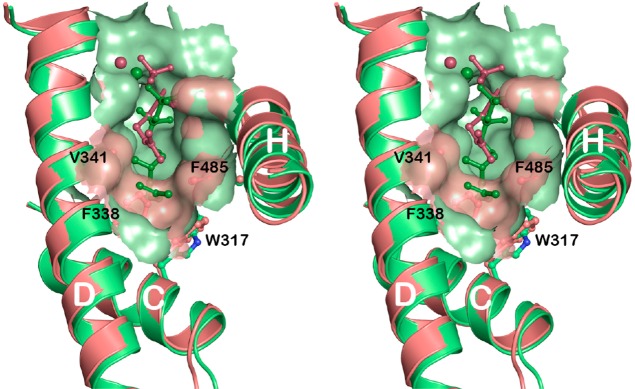
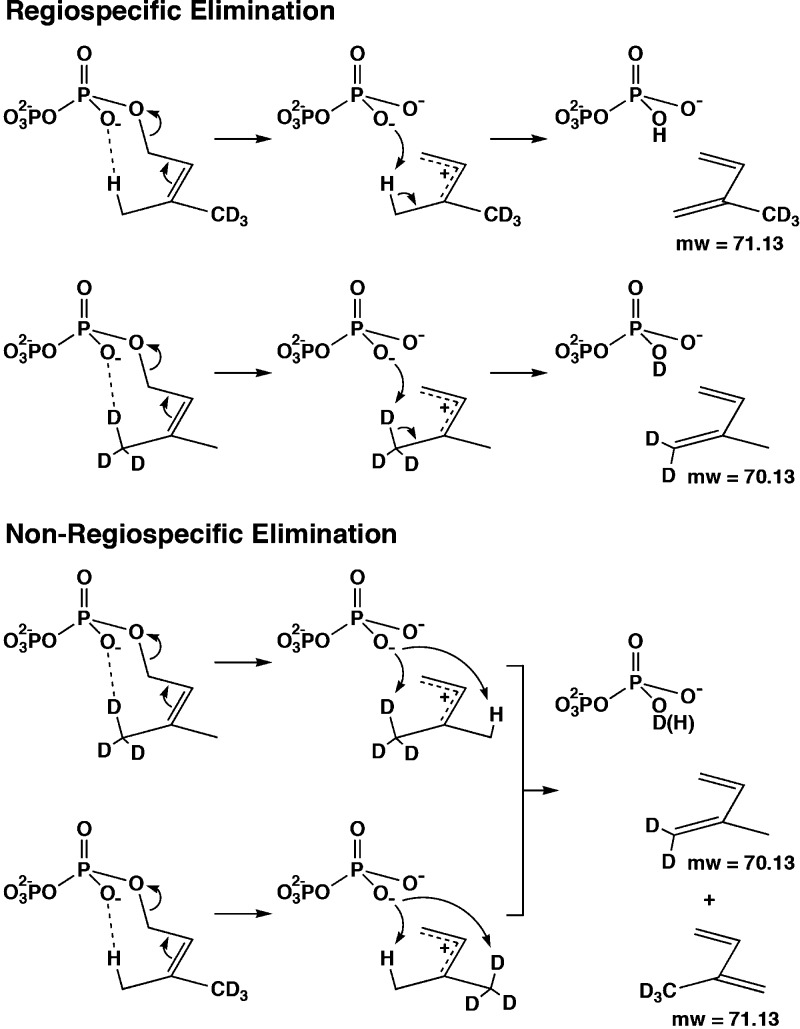
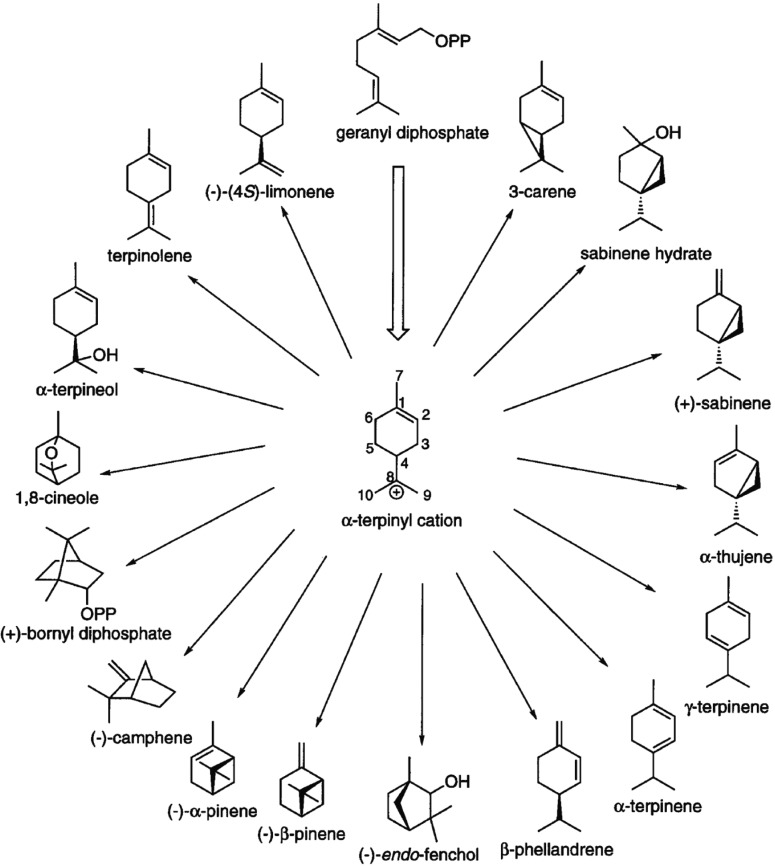
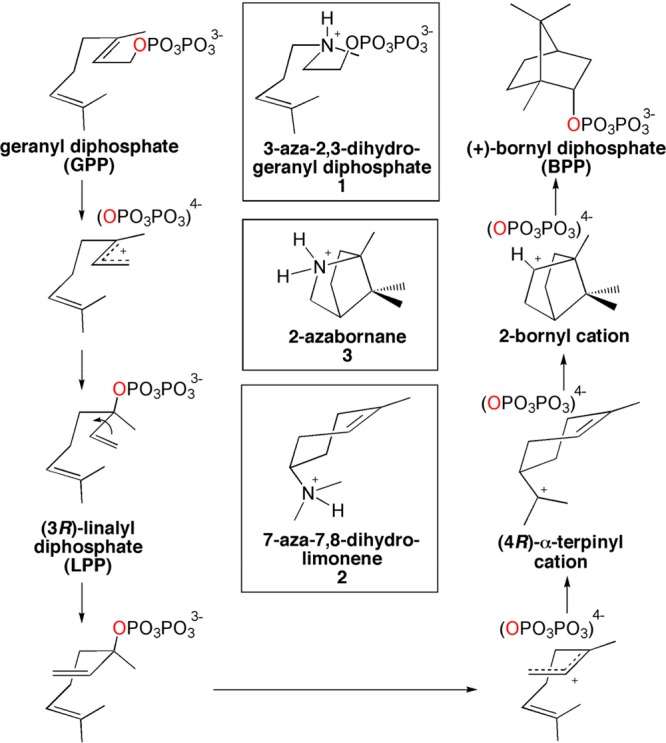
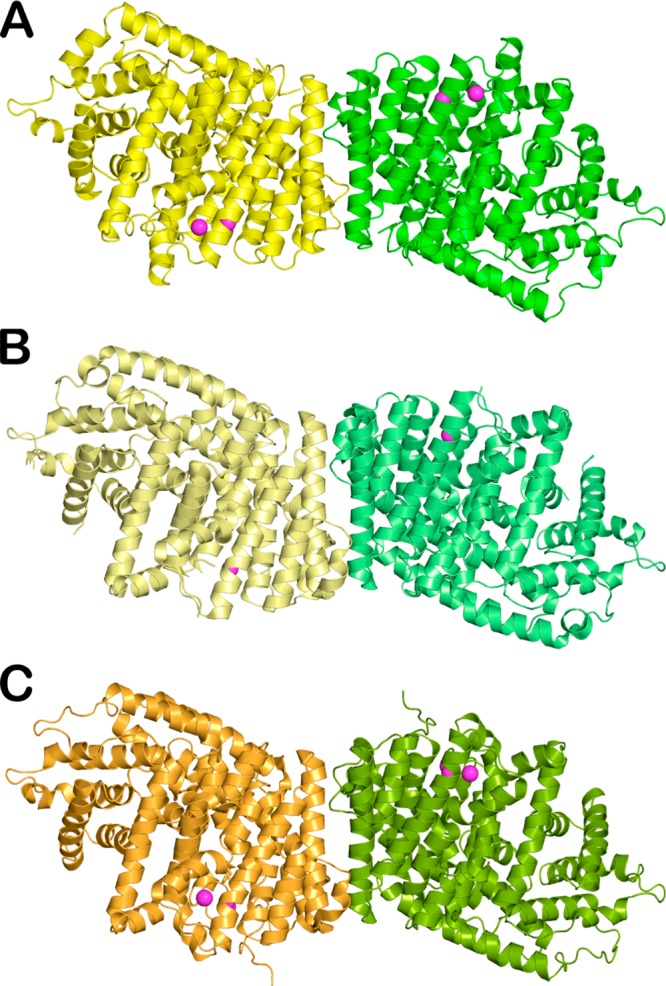
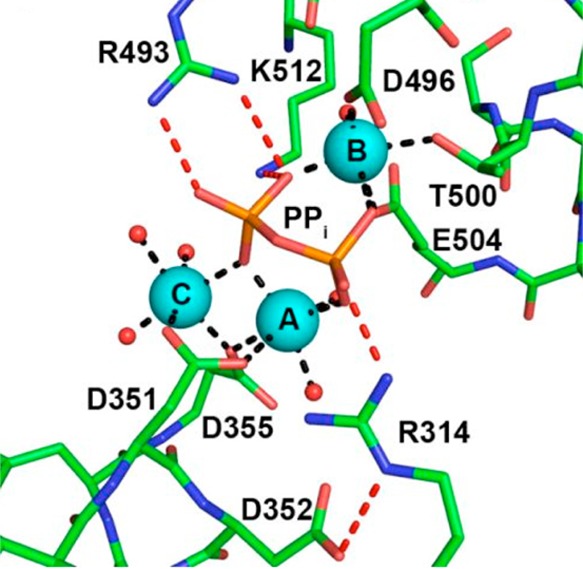
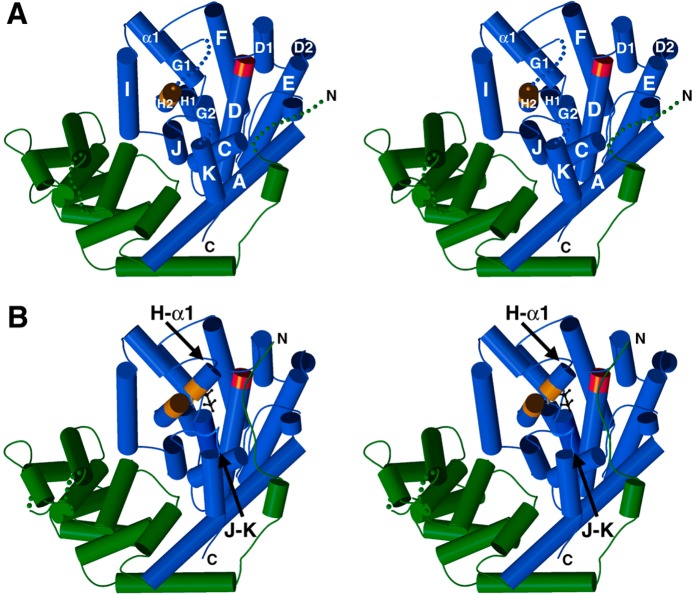
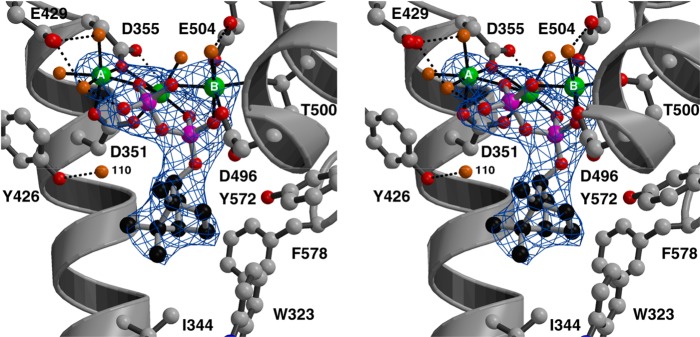
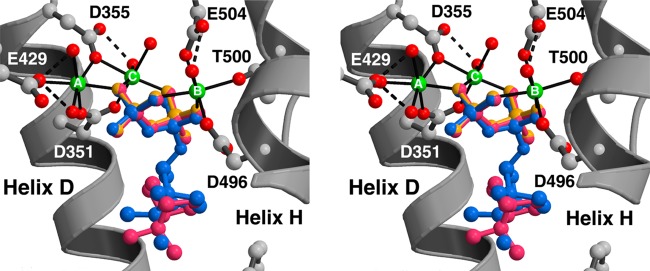
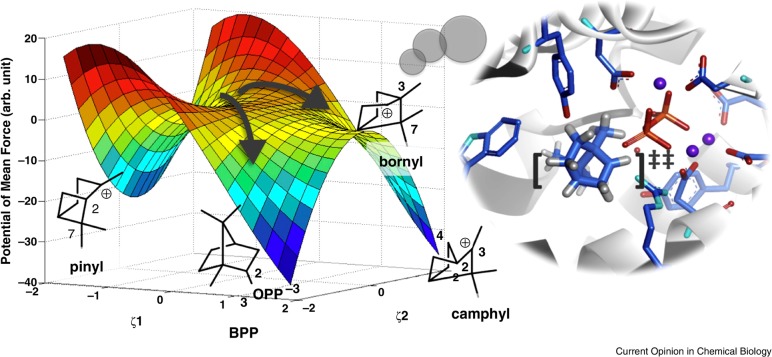
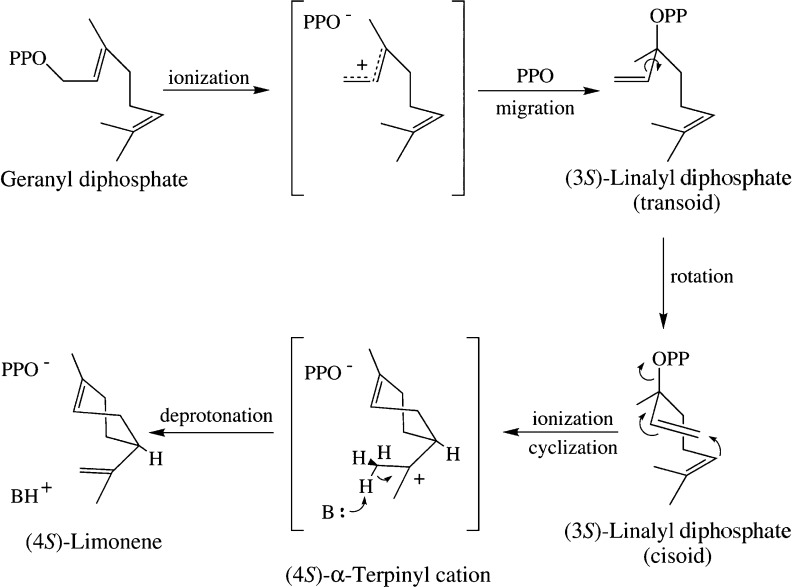
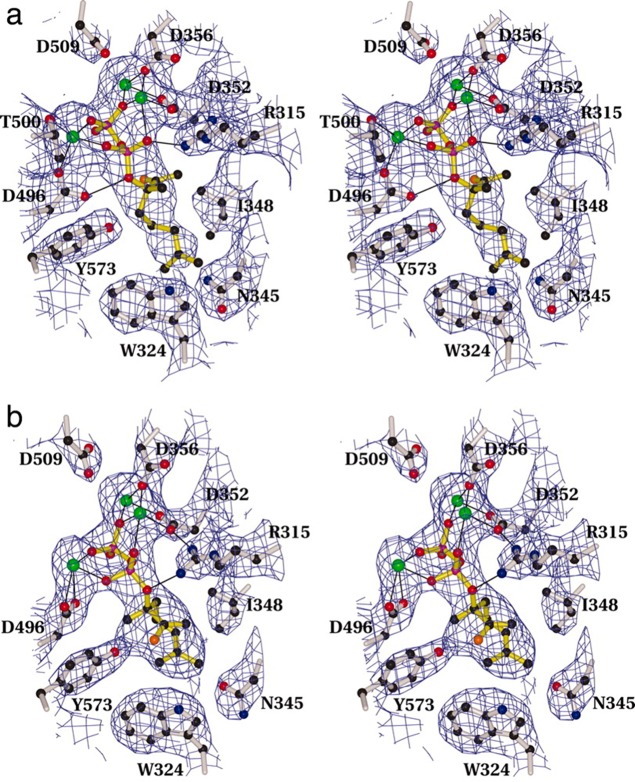
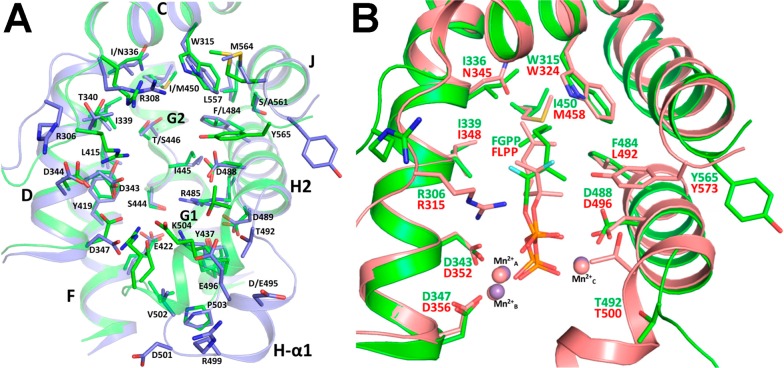
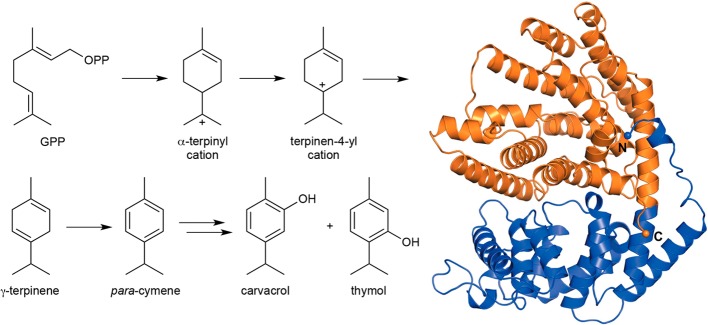
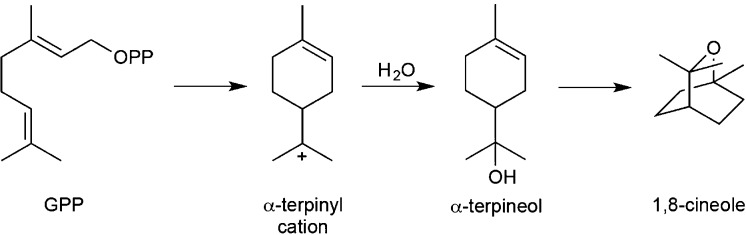
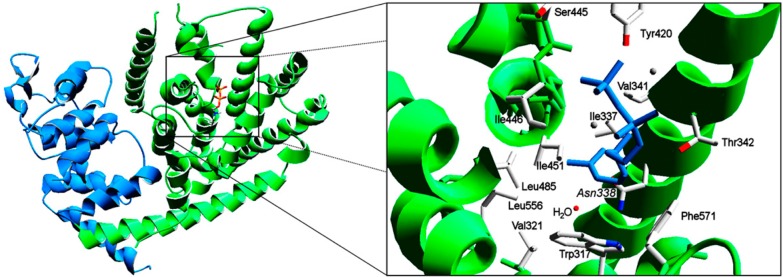
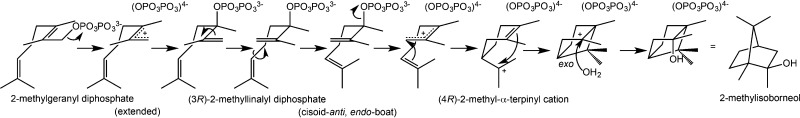
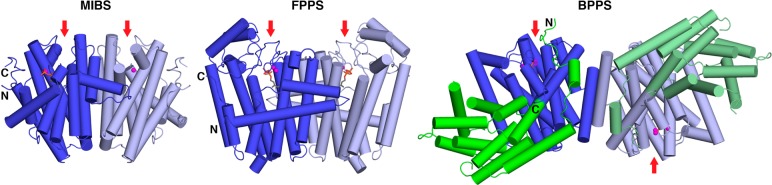
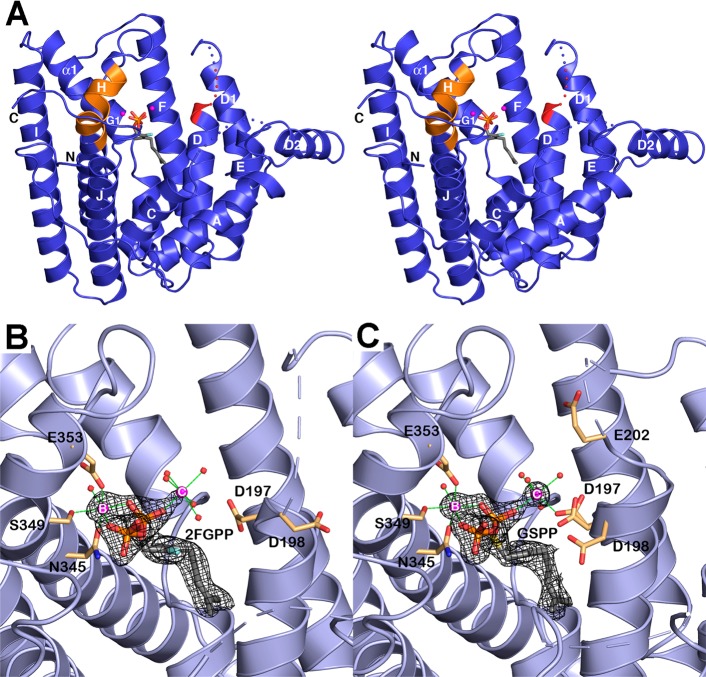
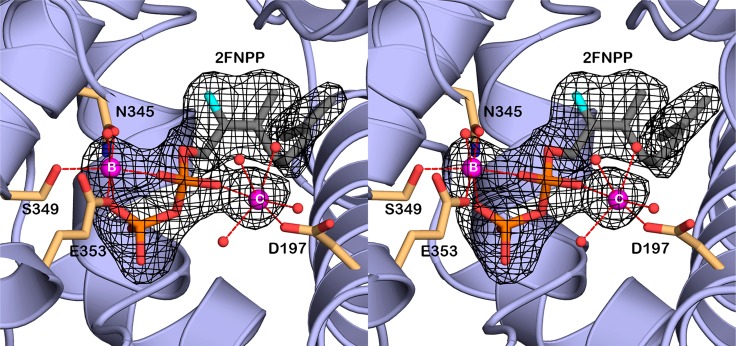
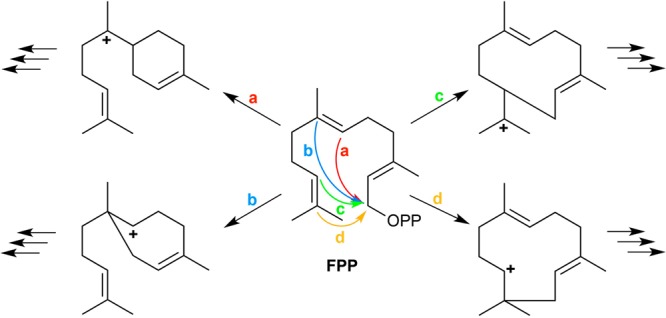
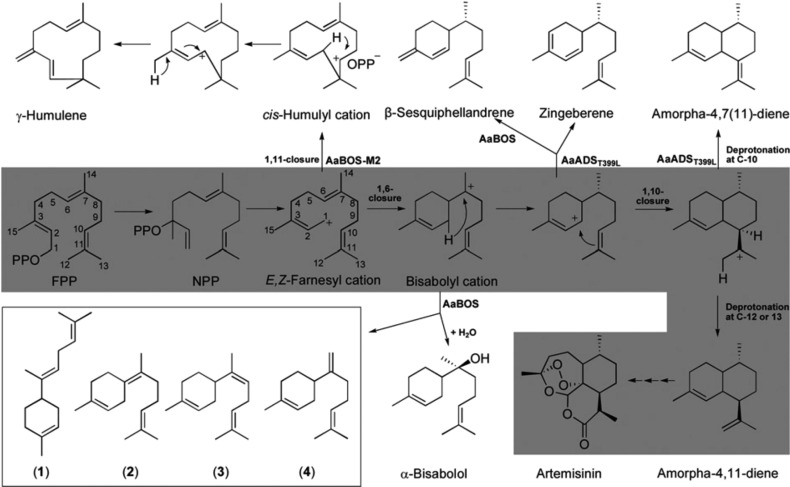
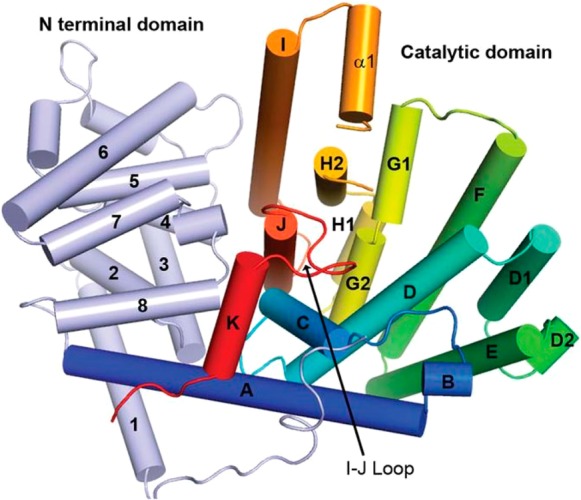
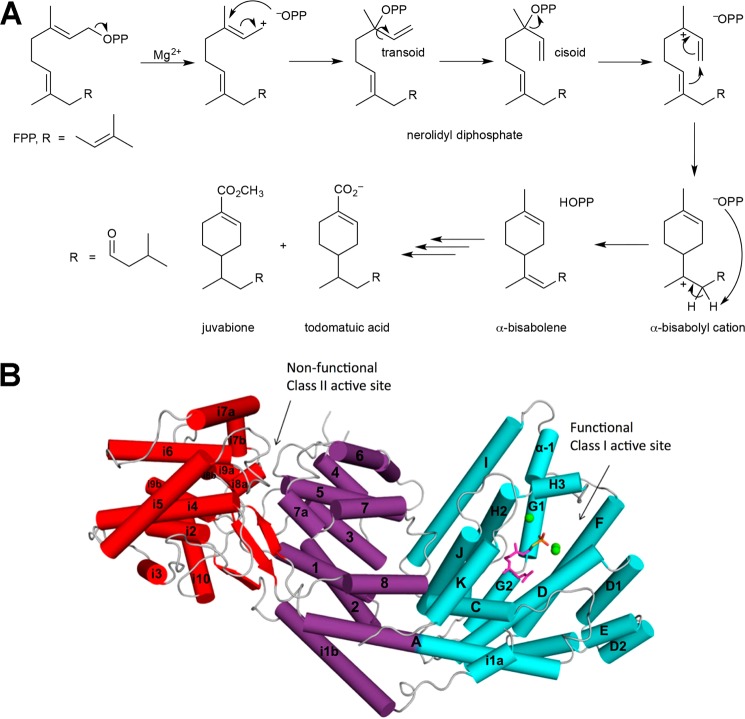
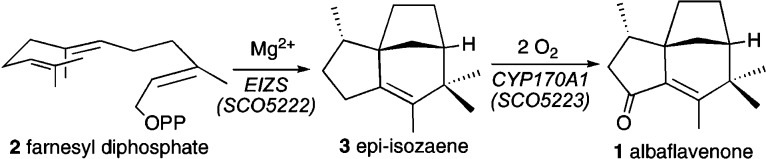
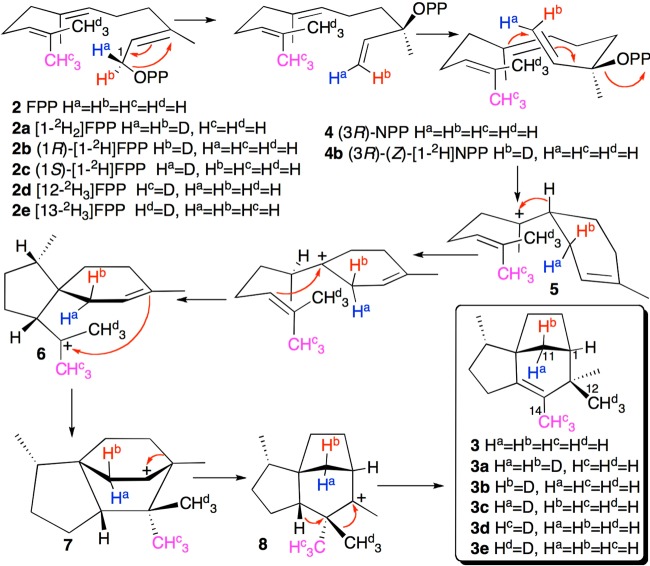
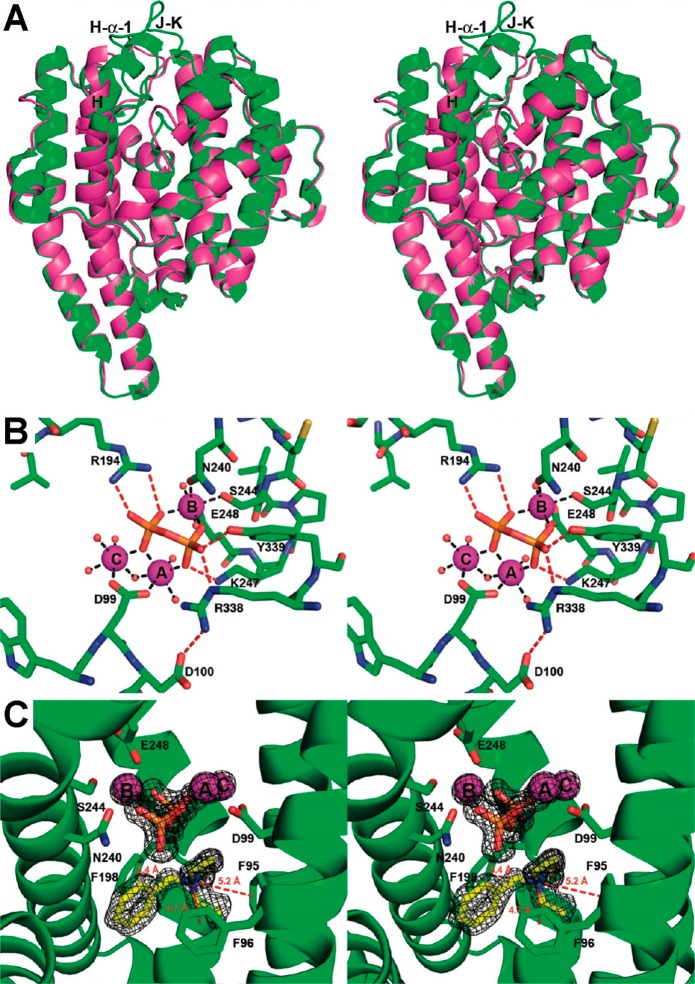
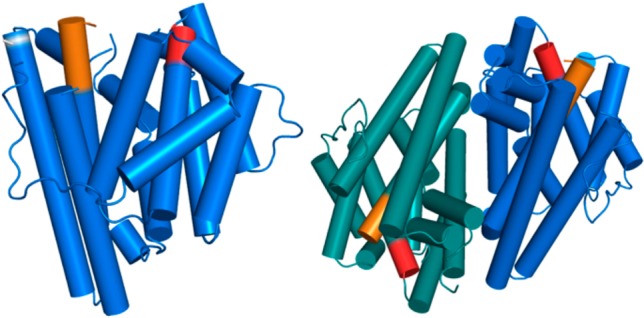
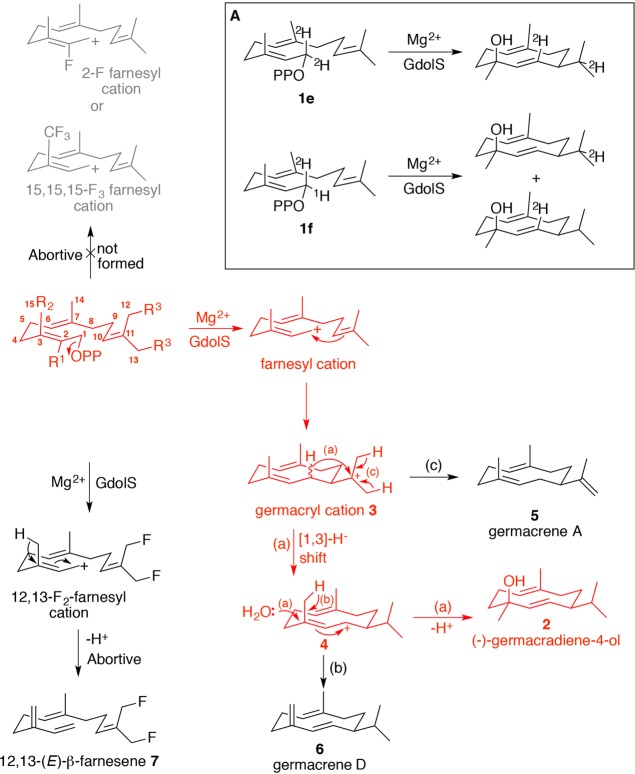
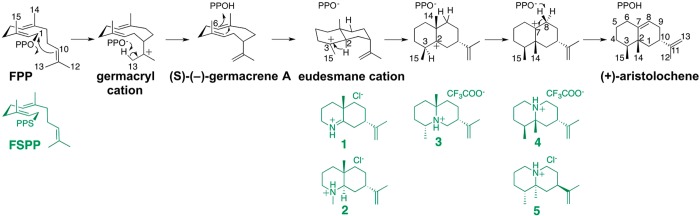
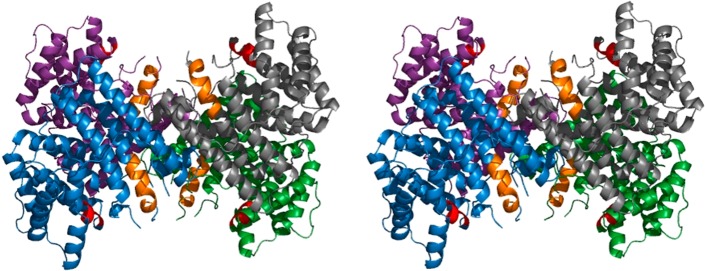
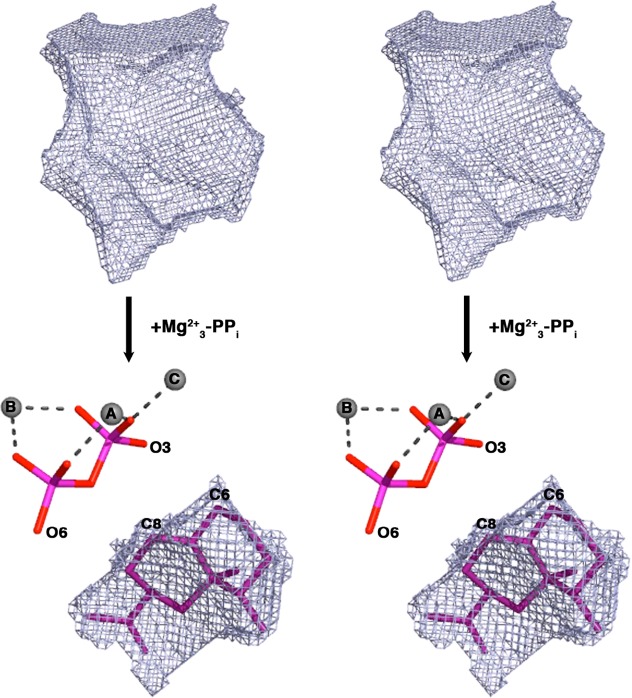
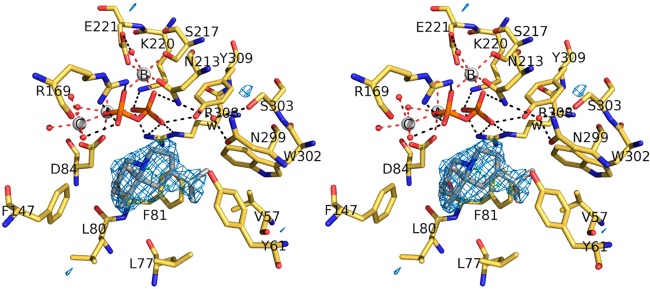
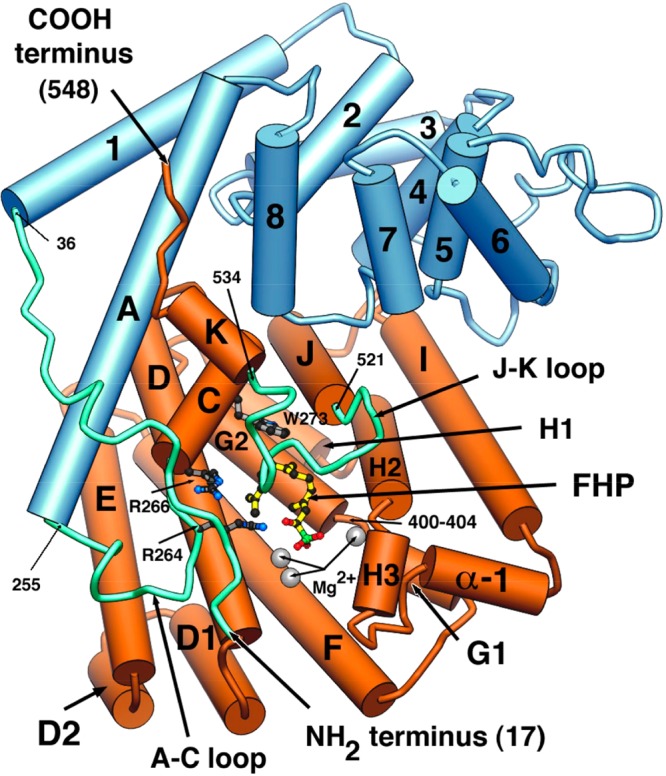
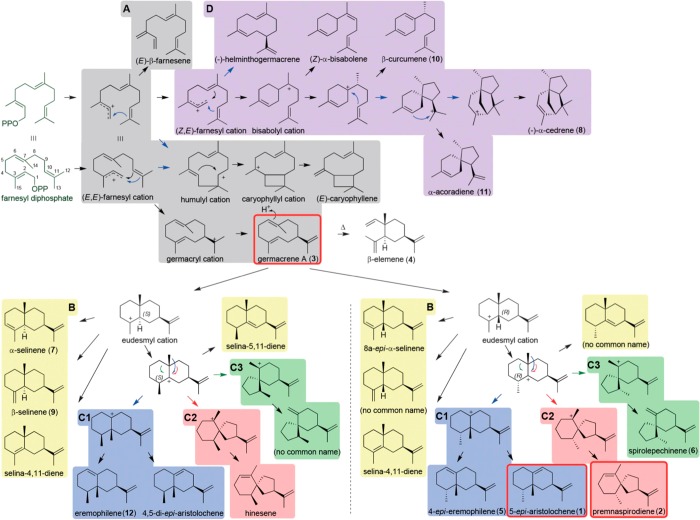
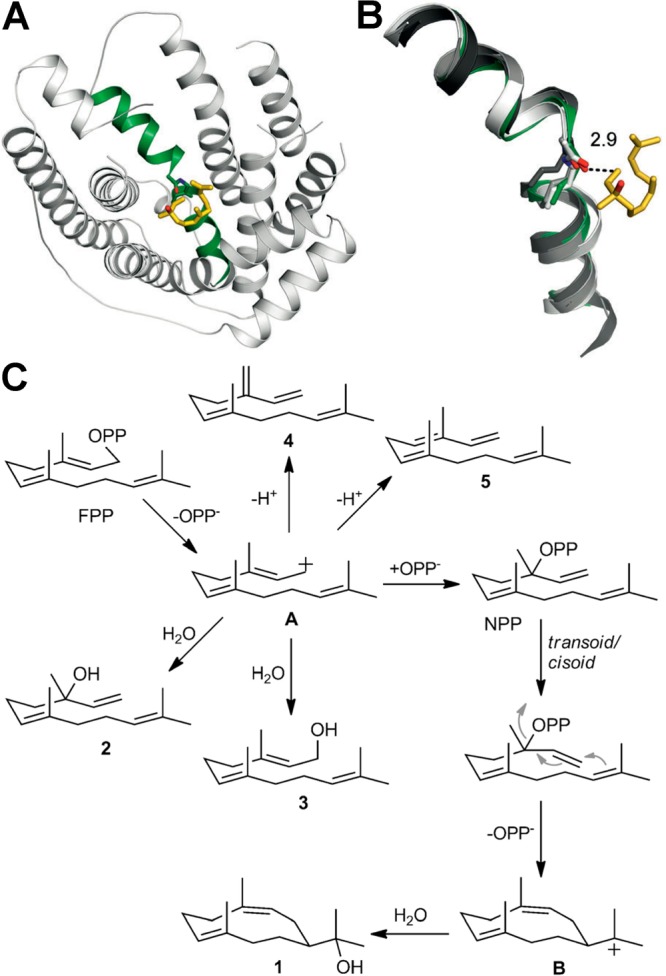
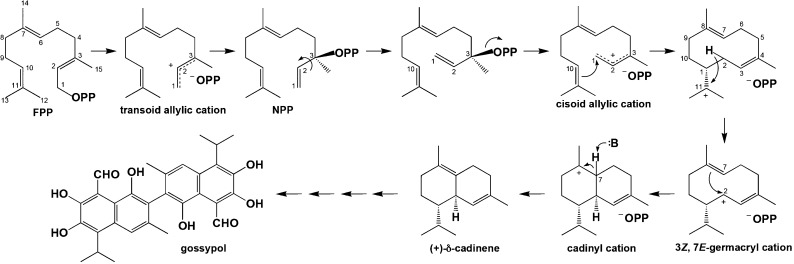
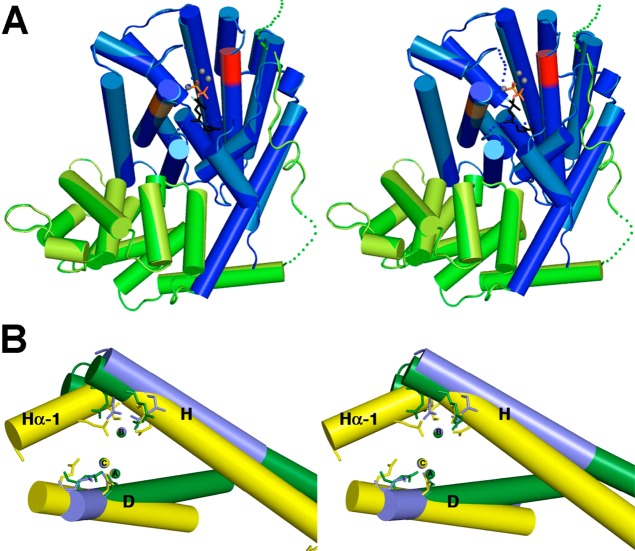
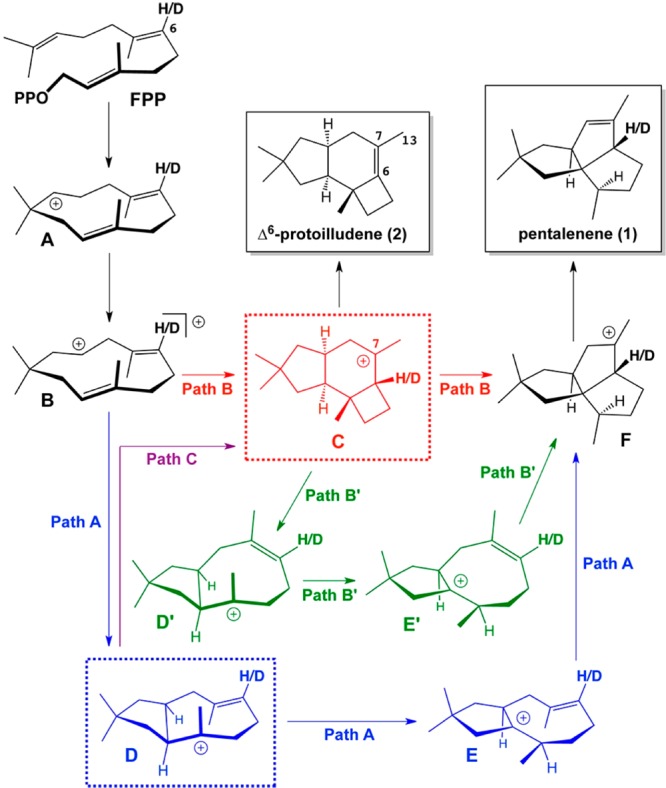
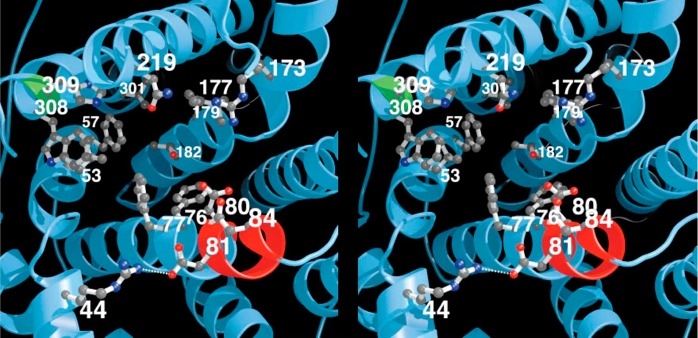

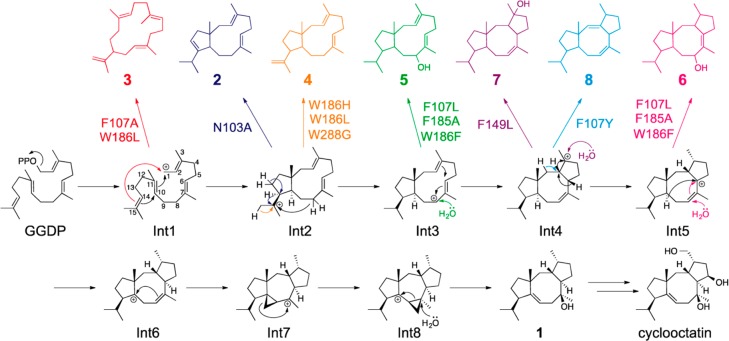
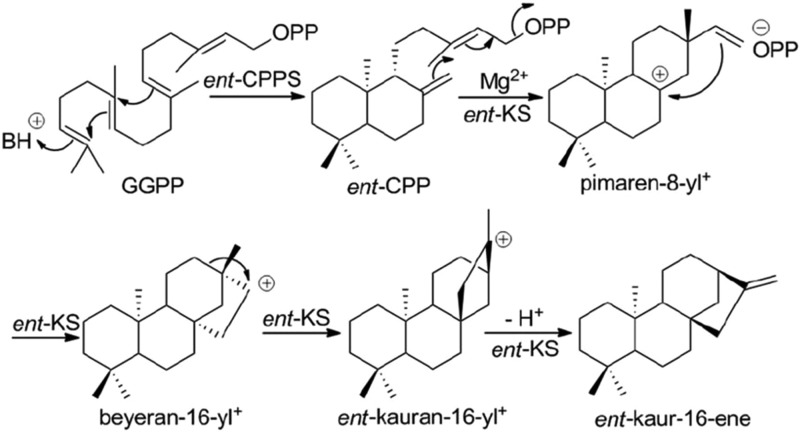
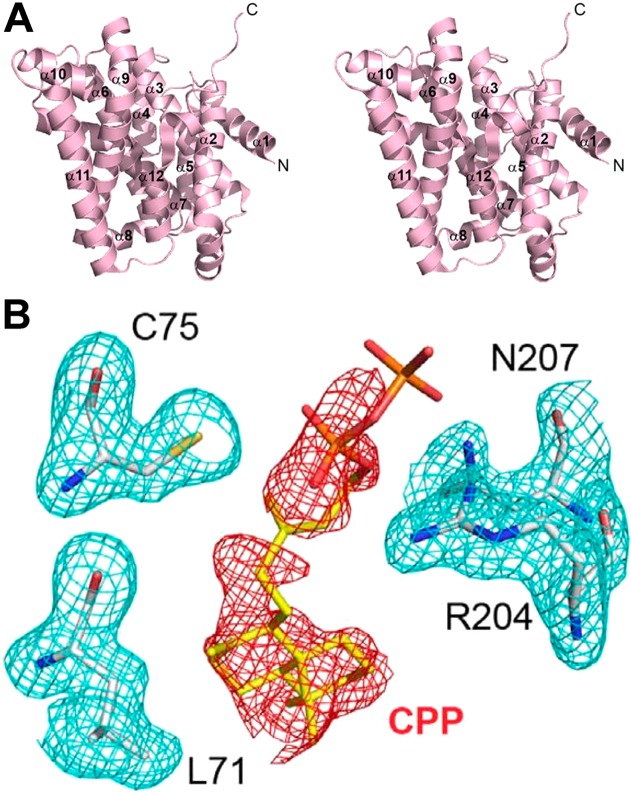
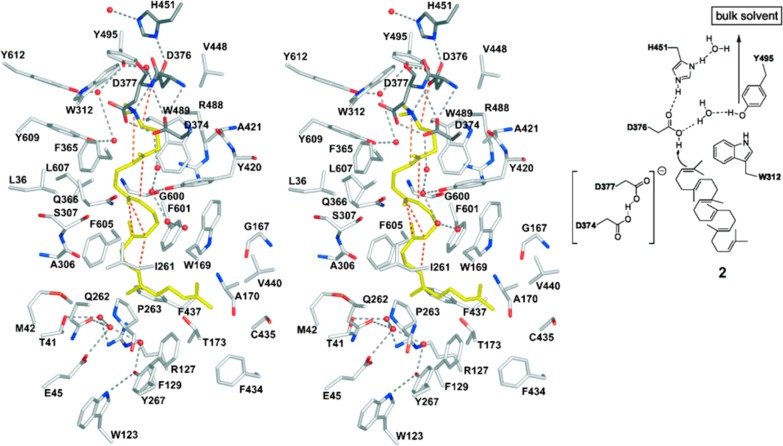
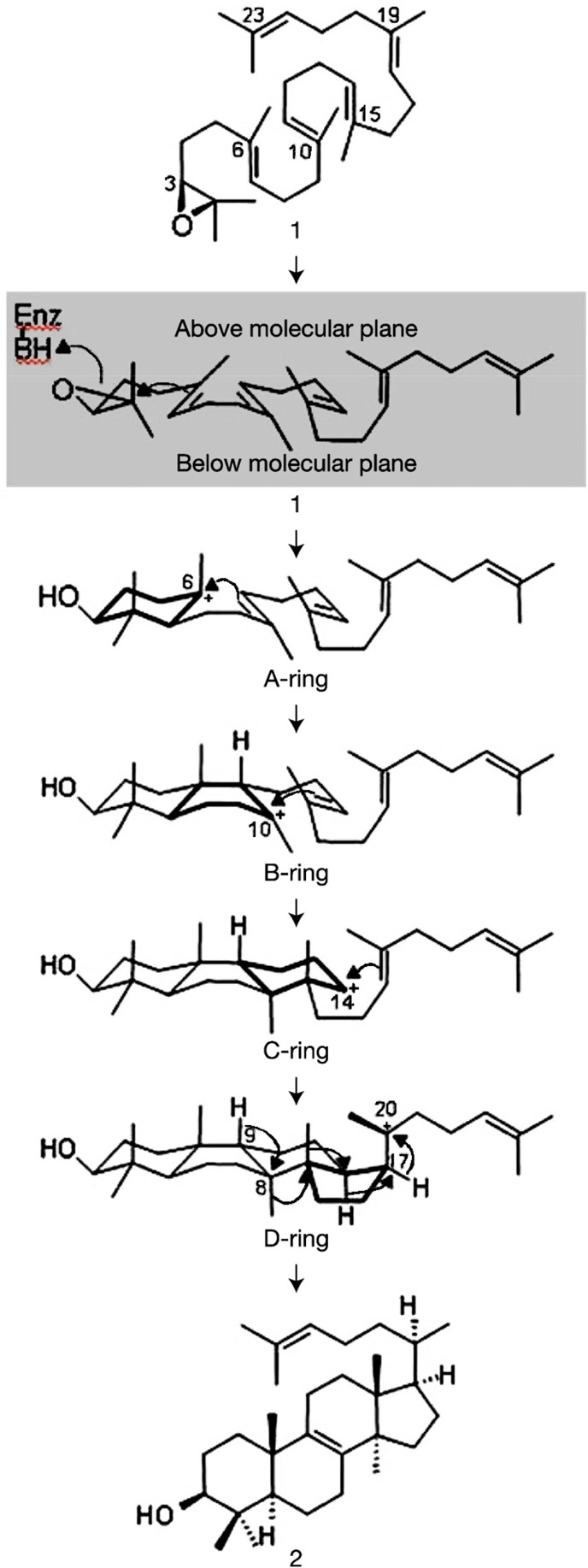
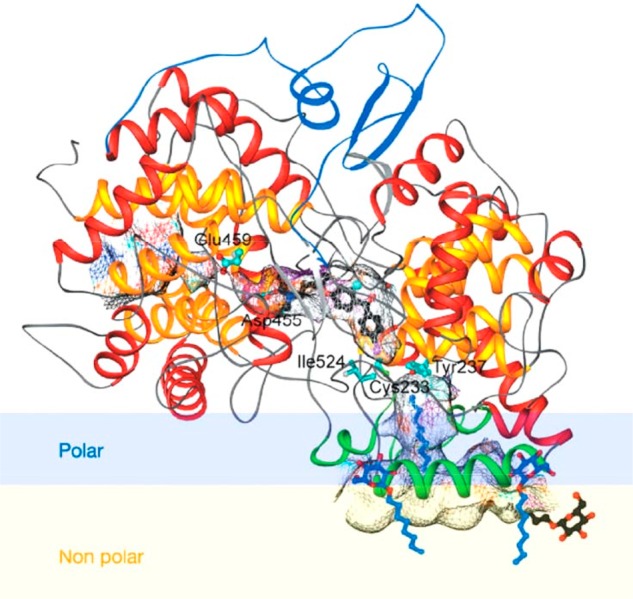
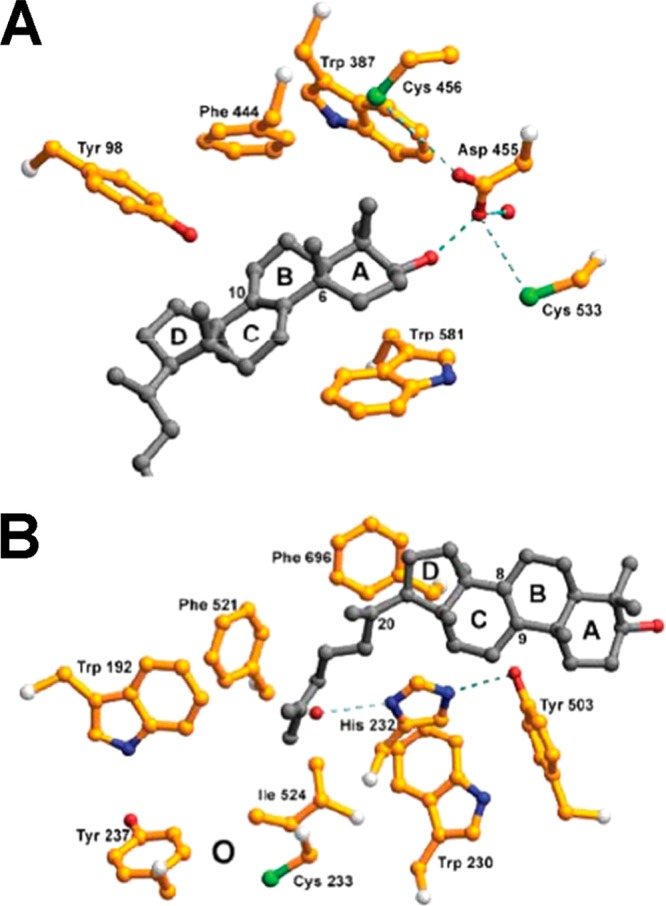
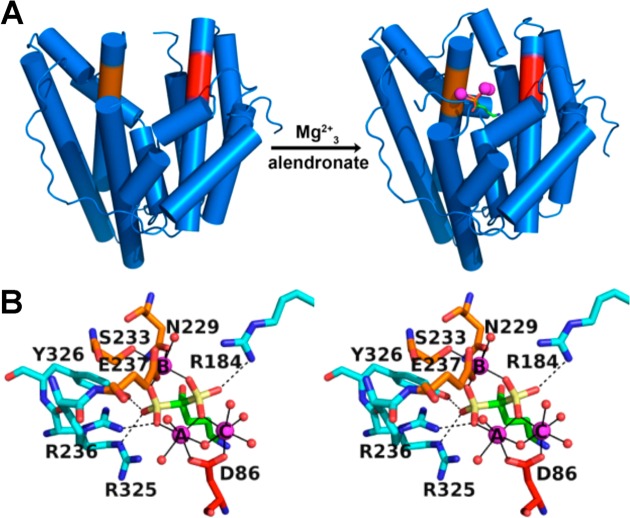
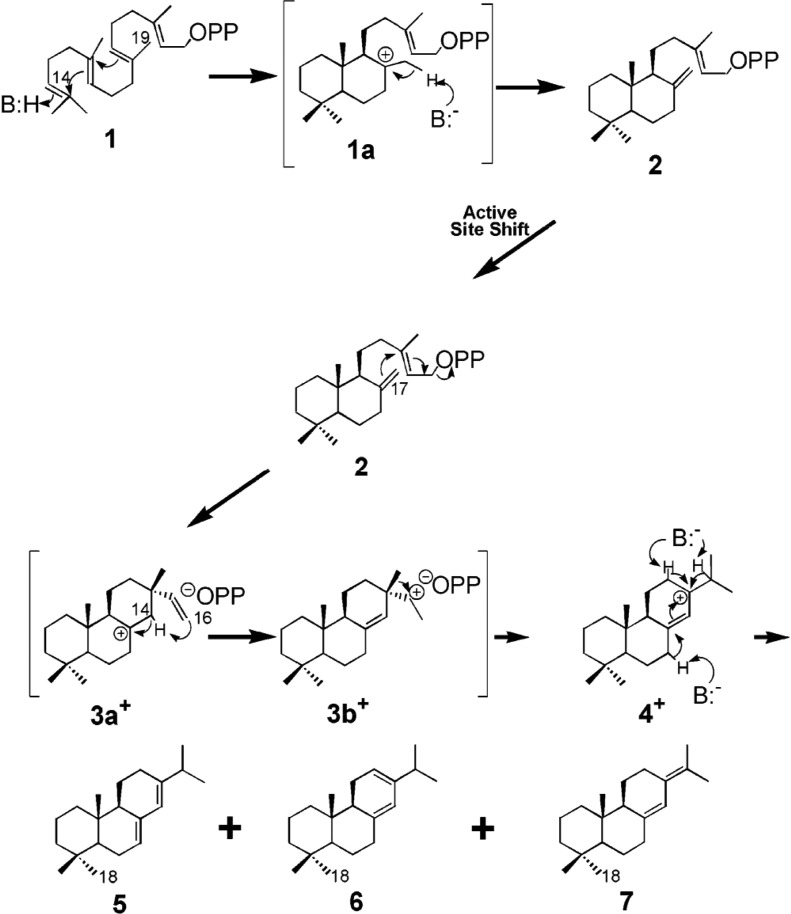
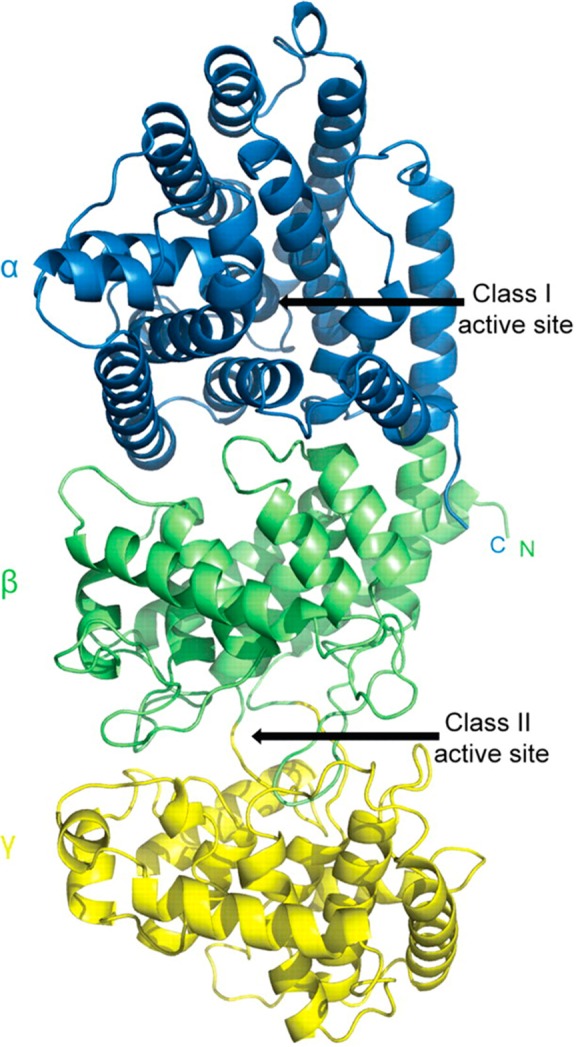
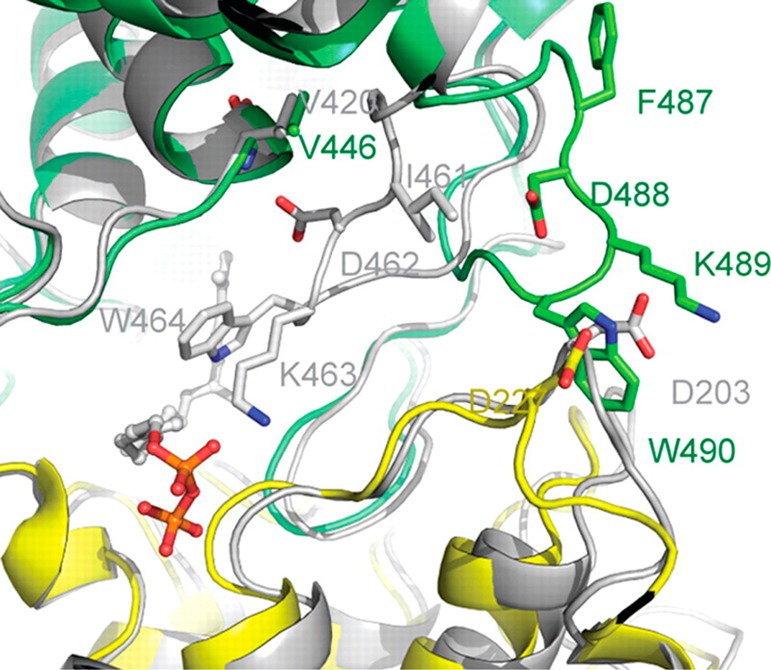
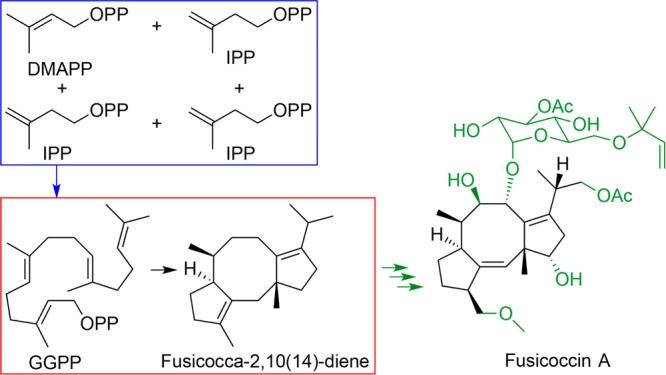
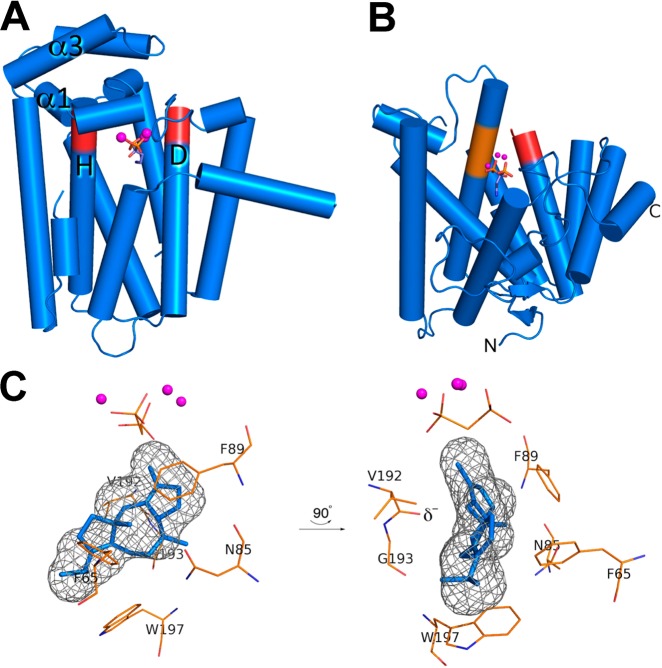
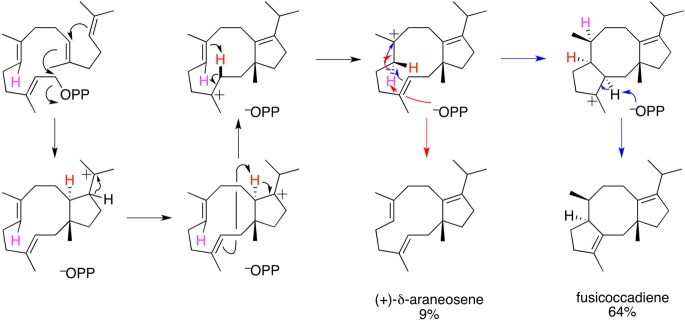
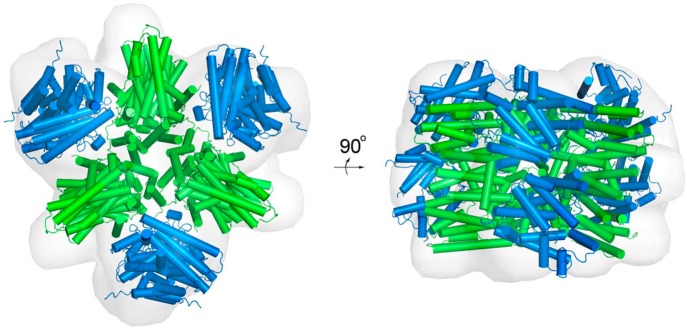
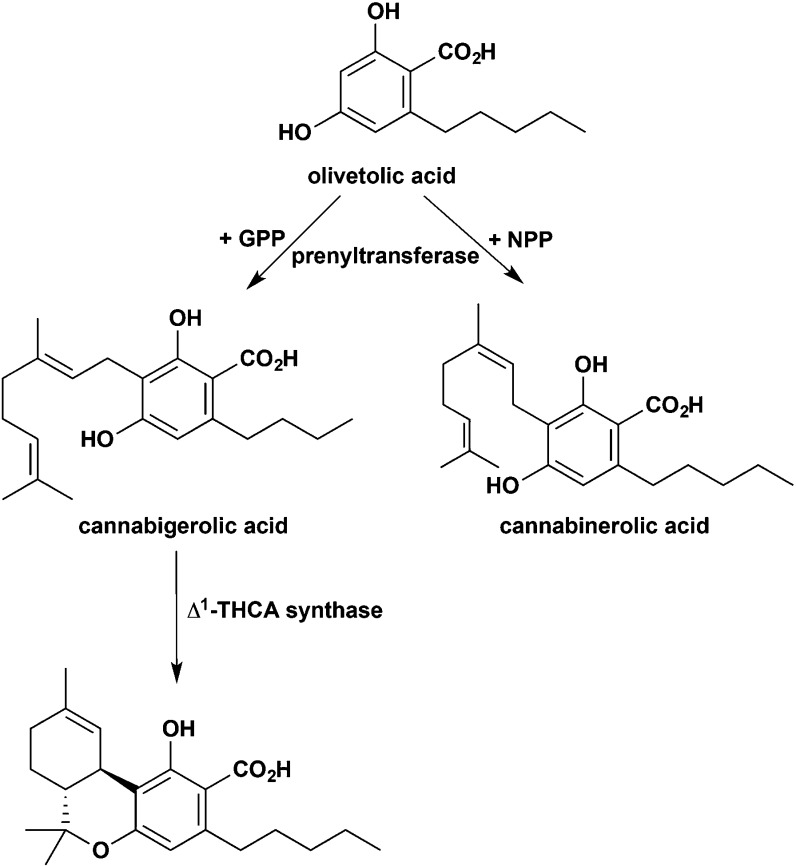
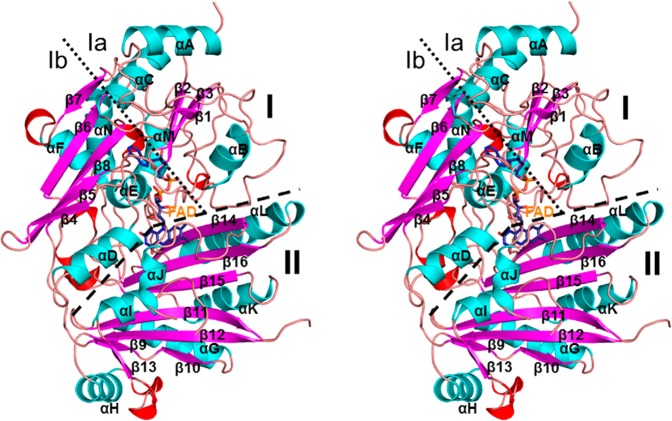
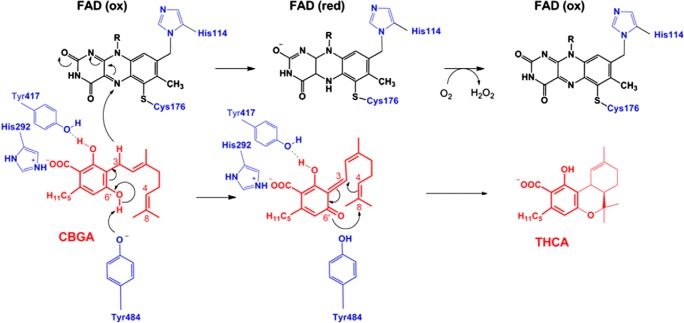
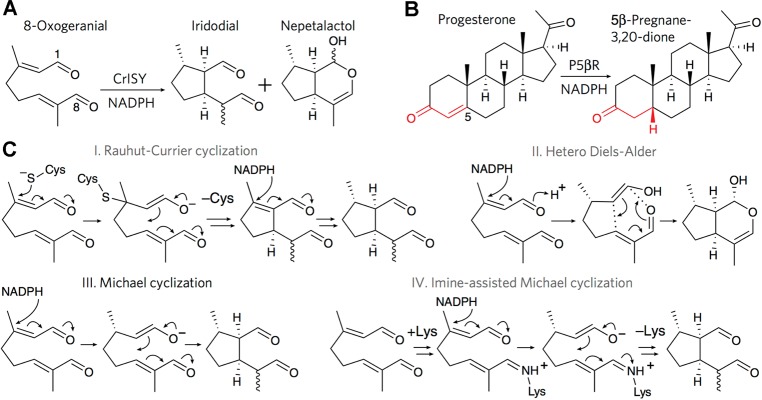
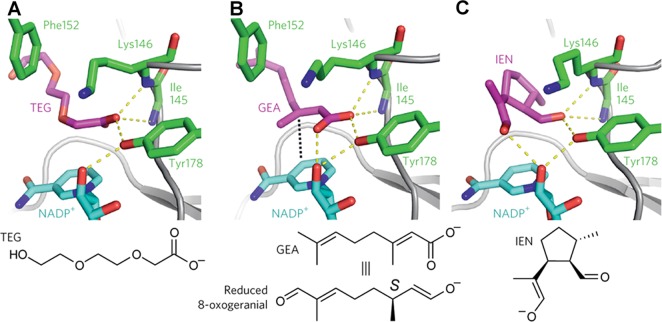
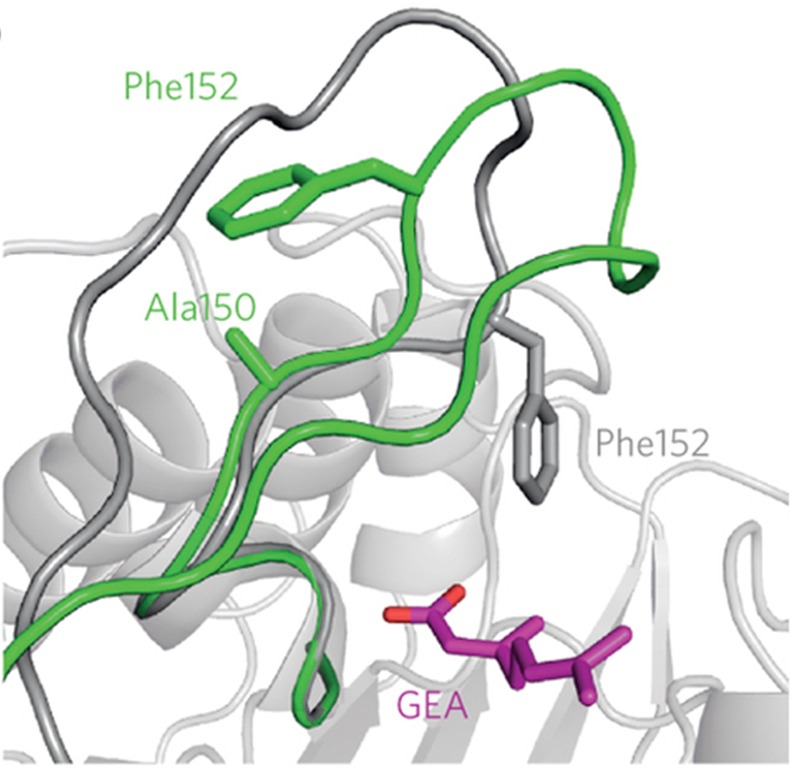
References
-
- Dictionary of Natural Products; Buckingham J., Ed.; Chapman & Hall, London, 1994; Vol. 7.
-
- Buckingham J.; Cooper C. M.; Purchase R.. Natural Products Desk Reference; CRC Press, Taylor & Francis Group: Boca Raton, 2016; p 235.
-
- Poulter C. D.; Rilling H. C. The Prenyl Transfer Reaction. Enzymatic and Mechanistic Studies of the 1′-4 Coupling Reaction in the Terpene Biosynthetic Pathway. Acc. Chem. Res. 1978, 11, 307–313. 10.1021/ar50128a004. - DOI
-
- Croteau R. Biosynthesis and Catabolism of Monoterpenoids. Chem. Rev. 1987, 87, 929–954. 10.1021/cr00081a004. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
