

Functional and molecular studies in primary carnitine deficiency
- PMID: 28841266
- PMCID: PMC5665702
- DOI: 10.1002/humu.23315
Functional and molecular studies in primary carnitine deficiency
Abstract
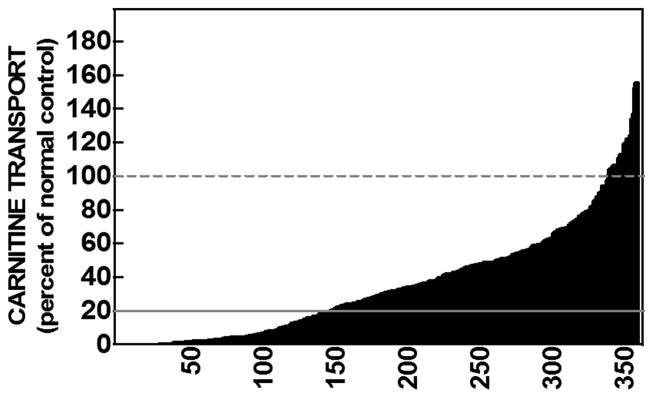
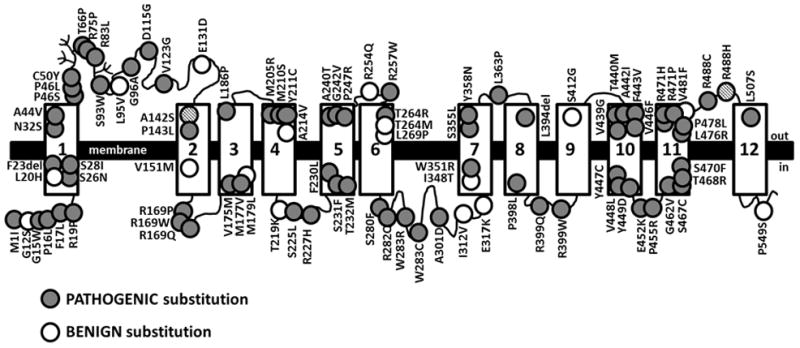
Primary carnitine deficiency is caused by a defect in the OCTN2 carnitine transporter encoded by the SLC22A5 gene. It can cause hypoketotic hypoglycemia or cardiomyopathy in children, and sudden death in children and adults. Fibroblasts from affected patients have reduced carnitine transport. We evaluated carnitine transport in fibroblasts from 358 subjects referred for possible carnitine deficiency. Carnitine transport was reduced to 20% or less of normal in fibroblasts of 140 out of 358 subjects. Sequencing of the 10 exons and flanking regions of the SLC22A5 gene in 95 out of 140 subjects identified causative variants in 84% of the alleles. The missense variants identified in our patients and others previously reported (n = 92) were expressed in CHO cells. Carnitine transport was impaired by 73 out of 92 variants expressed. Prediction algorithms (Polyphen-2, SIFT) correctly predicted the functional effects of expressed variants in about 80% of cases. These results indicate that mutations in the coding region of the SLC22A5 gene cannot be identified in about 16% of the alleles causing primary carnitine deficiency. Prediction algorithms failed to determine the functional effects of amino acid substitutions in this transmembrane protein in about 20% of cases. Therefore, functional studies in fibroblasts remain the best strategy to confirm or exclude a diagnosis of primary carnitine deficiency.
Keywords: OCTN2; SLC22A5; carnitine deficiency; carnitine transport; carnitine uptake defect; fatty acid oxidation; mutations; newborn screening.
© 2017 Wiley Periodicals, Inc.
Figures
References
-
- Akpinar B, Coteli C, Yucel D, Ozgul RK, Dursun A, Demirkol M, Baykal T, Gokcay G. A novel mutation (L363P) in the OCTN2 gene and its effect on secondary protein structure. Clinical Genetics. 2010;78(Suppl 1):89. (Abs J06)
-
- Amat di San Filippo C, Longo N. Tyrosine residues affecting sodium stimulation of carnitine transport in the OCTN2 carnitine/organic cation transporter. J Biol Chem. 2004;279(8):7247–53. - PubMed
-
- Amat di San Filippo C, Pasquali M, Longo N. Pharmacological rescue of carnitine transport in primary carnitine deficiency. Hum Mutat. 2006;27(6):513–23. - PubMed
-
- Amat di San Filippo C, Wang Y, Longo N. Functional domains in the carnitine transporter OCTN2, defective in primary carnitine deficiency. J Biol Chem. 2003;278(48):47776–84. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
