

A natural history study of X-linked myotubular myopathy
- PMID: 28842446
- PMCID: PMC5649758
- DOI: 10.1212/WNL.0000000000004415
A natural history study of X-linked myotubular myopathy
Abstract
Objective: To define the natural history of X-linked myotubular myopathy (MTM).
Methods: We performed a cross-sectional study that included an online survey (n = 35) and a prospective, 1-year longitudinal investigation using a phone survey (n = 33).
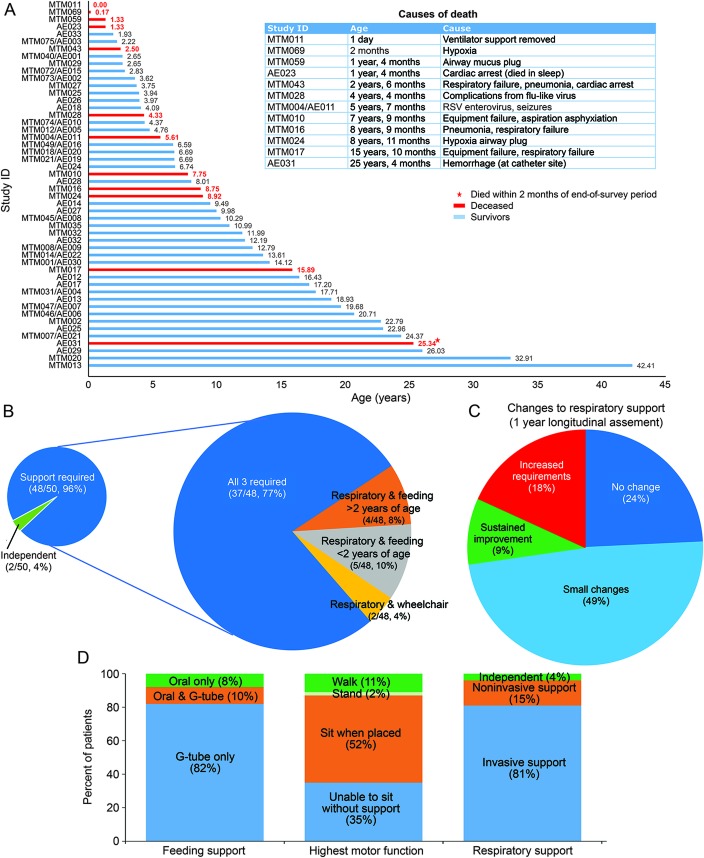
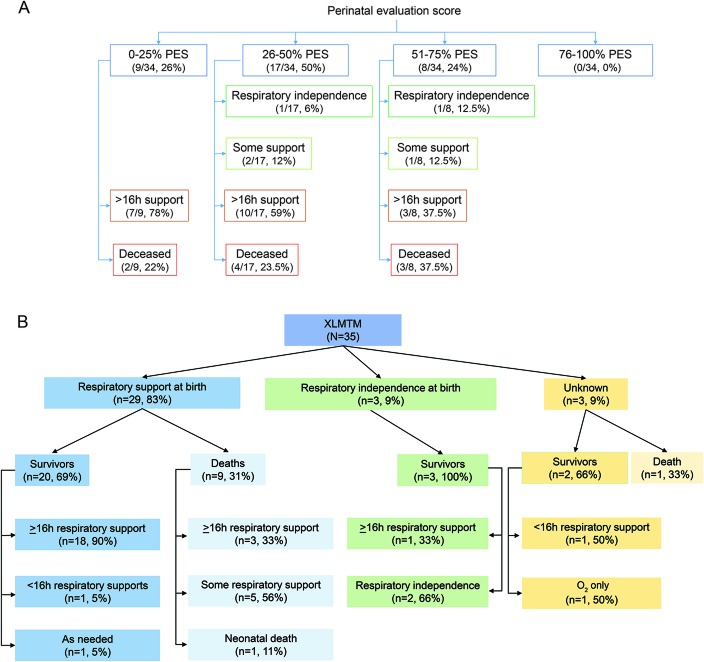
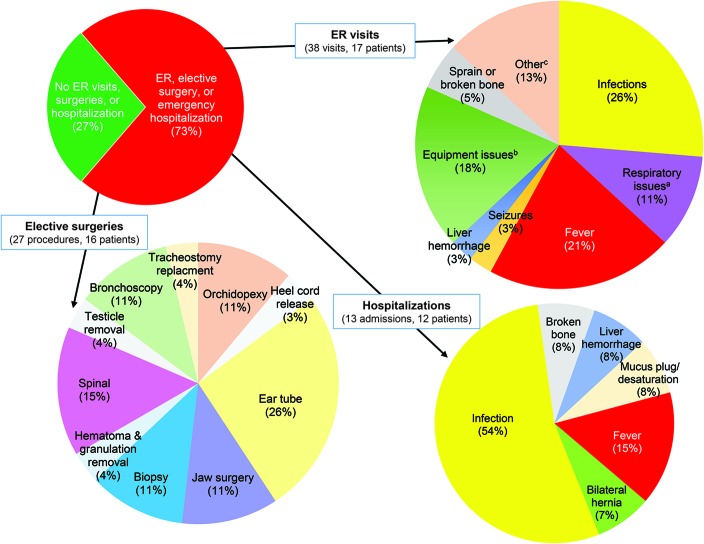
Results: We ascertained data from 50 male patients with MTM and performed longitudinal assessments on 33 affected individuals. Consistent with existing knowledge, we found that MTM is a disorder associated with extensive morbidities, including wheelchair (86.7% nonambulant) and ventilator (75% requiring >16 hours of support) dependence. However, unlike previous reports and despite the high burden of disease, mortality was lower than anticipated (approximate rate 10%/y). Seventy-six percent of patients with MTM enrolled (mean age 10 years 11 months) were alive at the end of the study. Nearly all deaths in the study were associated with respiratory failure. In addition, the disease course was more stable than expected, with few adverse events reported during the prospective survey. Few non-muscle-related morbidities were identified, although an unexpectedly high incidence of learning disability (43%) was noted. Conversely, MTM was associated with substantial burdens on patient and caregiver daily living, reflected by missed days of school and lost workdays.
Conclusions: MTM is one of the most severe neuromuscular disorders, with affected individuals requiring extensive mechanical interventions for survival. However, among study participants, the disease course was more stable than predicted, with more individuals surviving infancy and early childhood. These data reflect the disease burden of MTM but offer hope in terms of future therapeutic intervention.
Copyright © 2017 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures

Comment in
-
X-linked myotubular myopathy: Living longer and awaiting treatment.Neurology. 2017 Sep 26;89(13):1316-1317. doi: 10.1212/WNL.0000000000004428. Epub 2017 Aug 25. Neurology. 2017. PMID: 28842443 No abstract available.
References
-
- Laporte J, Hu LJ, Kretz C, et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet 1996;13:175–182. - PubMed
-
- Das S, Dowling J, Pierson CR. X-linked centronuclear myopathy. In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews. Seattle, WA: University of Washington; 1993–2017.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical