

5-hydroxymethylcytosine accumulation in postmitotic neurons results in functional demethylation of expressed genes
- PMID: 28847947
- PMCID: PMC5604027
- DOI: 10.1073/pnas.1708044114
5-hydroxymethylcytosine accumulation in postmitotic neurons results in functional demethylation of expressed genes
Abstract
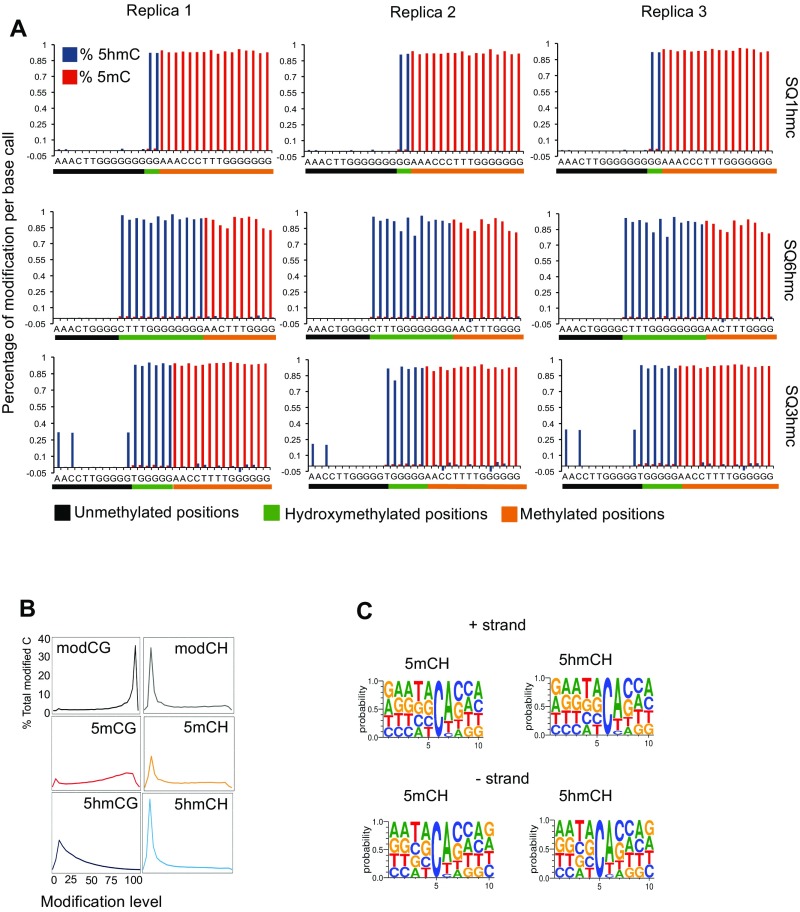
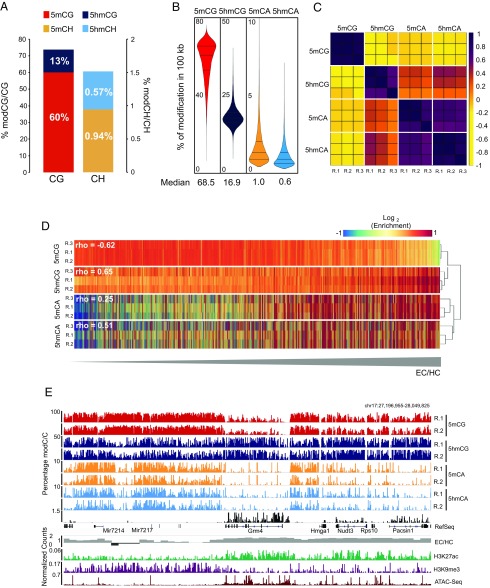
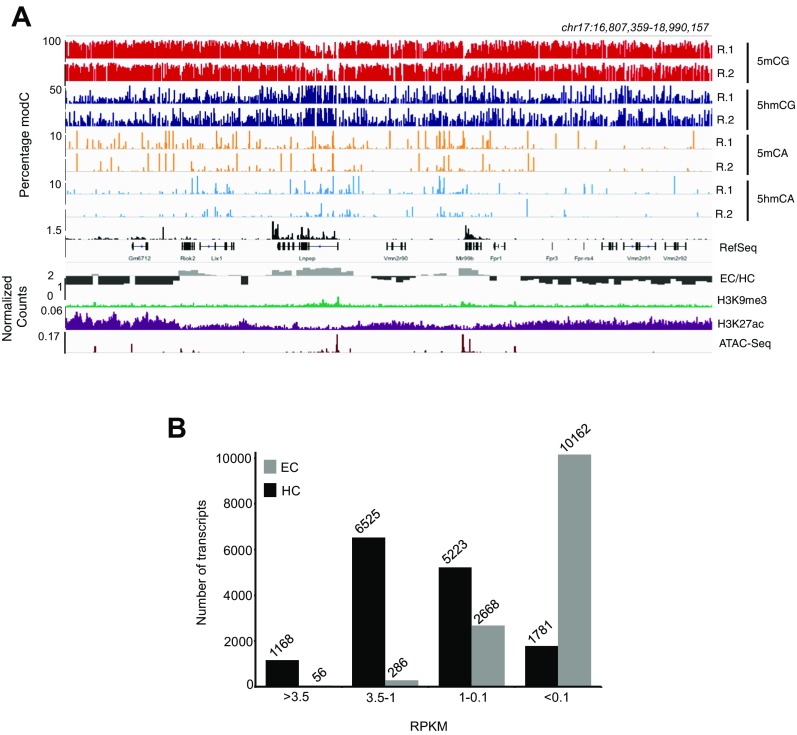
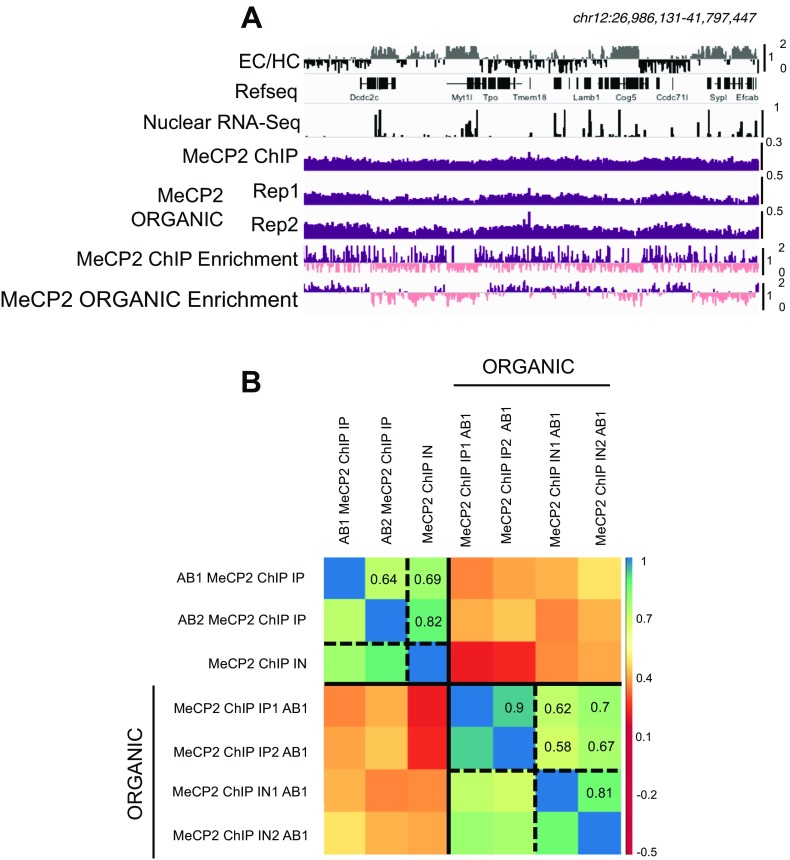
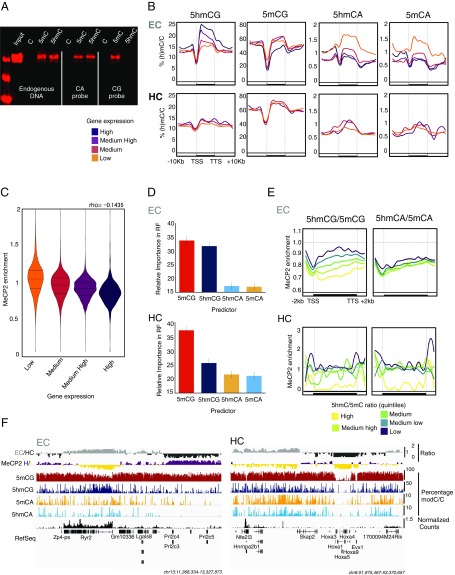
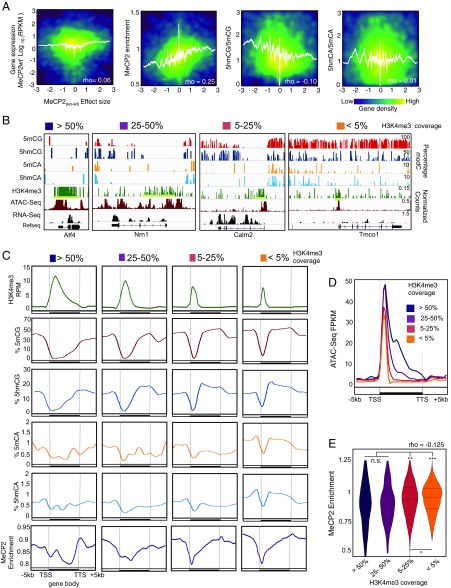
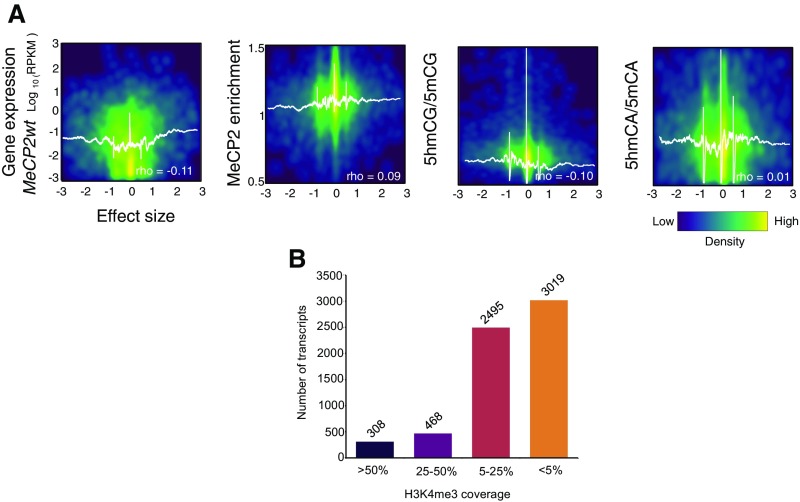
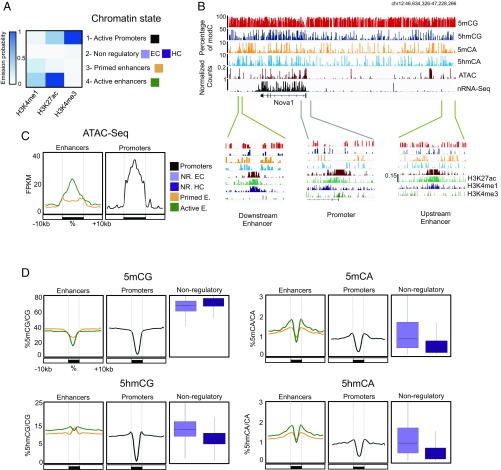
5-hydroxymethylcytosine (5hmC) occurs at maximal levels in postmitotic neurons, where its accumulation is cell-specific and correlated with gene expression. Here we demonstrate that the distribution of 5hmC in CG and non-CG dinucleotides is distinct and that it reflects the binding specificity and genome occupancy of methylcytosine binding protein 2 (MeCP2). In expressed gene bodies, accumulation of 5hmCG acts in opposition to 5mCG, resulting in "functional" demethylation and diminished MeCP2 binding, thus facilitating transcription. Non-CG hydroxymethylation occurs predominantly in CA dinucleotides (5hmCA) and it accumulates in regions flanking active enhancers. In these domains, oxidation of 5mCA to 5hmCA does not alter MeCP2 binding or expression of adjacent genes. We conclude that the role of 5-hydroxymethylcytosine in postmitotic neurons is to functionally demethylate expressed gene bodies while retaining the role of MeCP2 in chromatin organization.
Keywords: 5-hydroxymethylcytosine; MeCP2; epigenetics; neuron.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
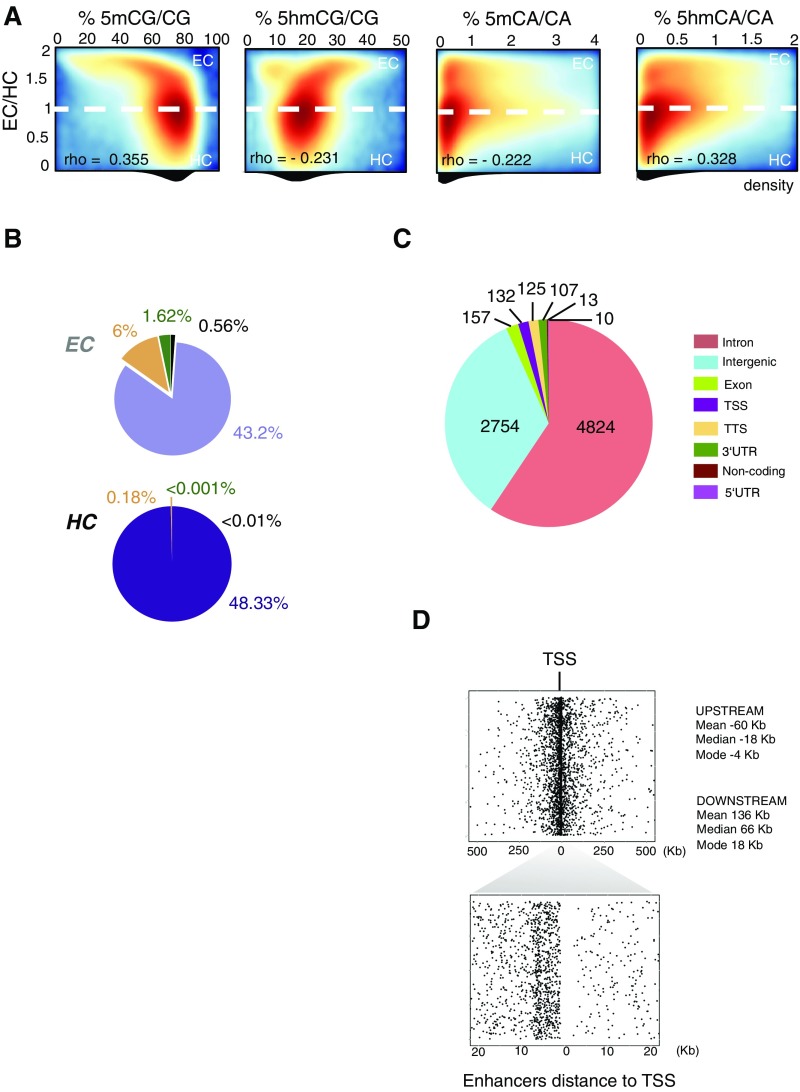
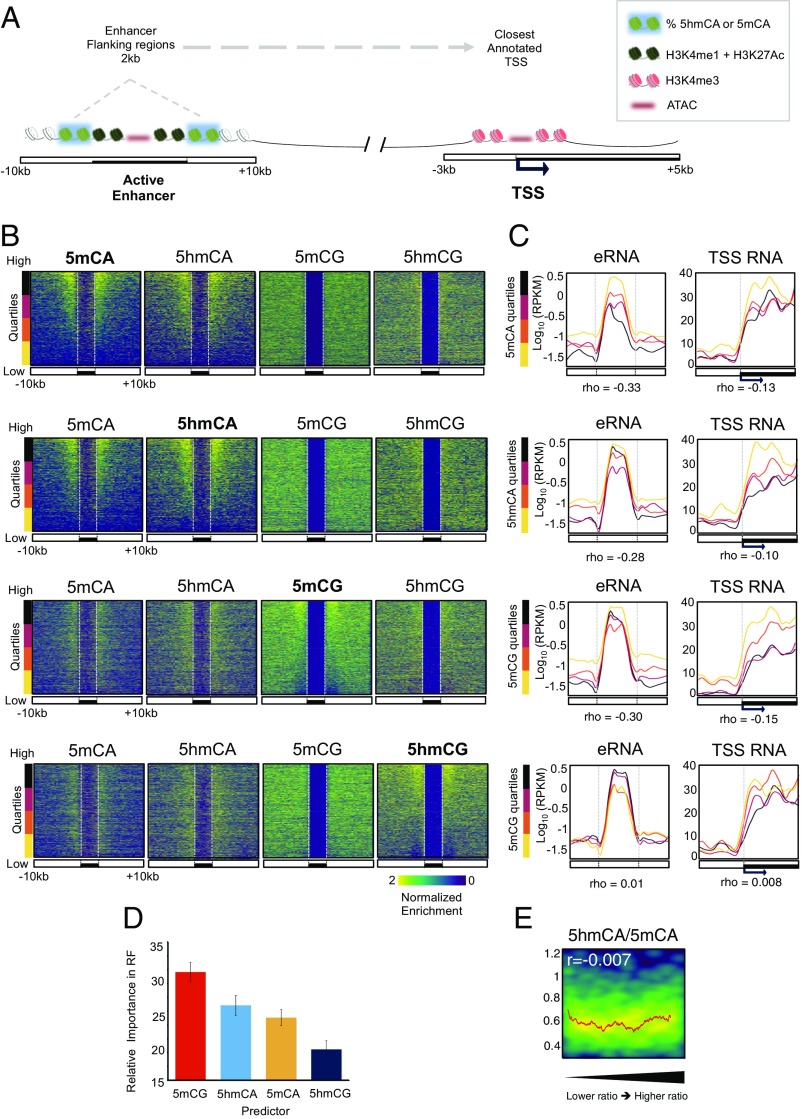
References
-
- Suzuki MM, Bird A. DNA methylation landscapes: Provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
