

TRP53 Mutants Drive Neuroendocrine Lung Cancer Through Loss-of-Function Mechanisms with Gain-of-Function Effects on Chemotherapy Response
- PMID: 28847987
- PMCID: PMC5716875
- DOI: 10.1158/1535-7163.MCT-17-0353
TRP53 Mutants Drive Neuroendocrine Lung Cancer Through Loss-of-Function Mechanisms with Gain-of-Function Effects on Chemotherapy Response
Abstract
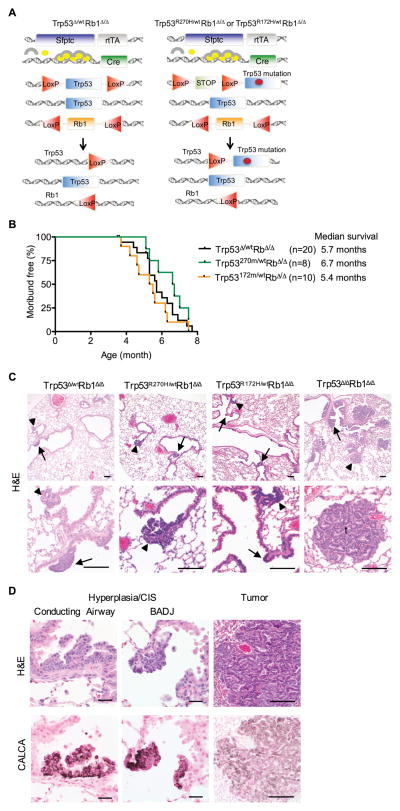
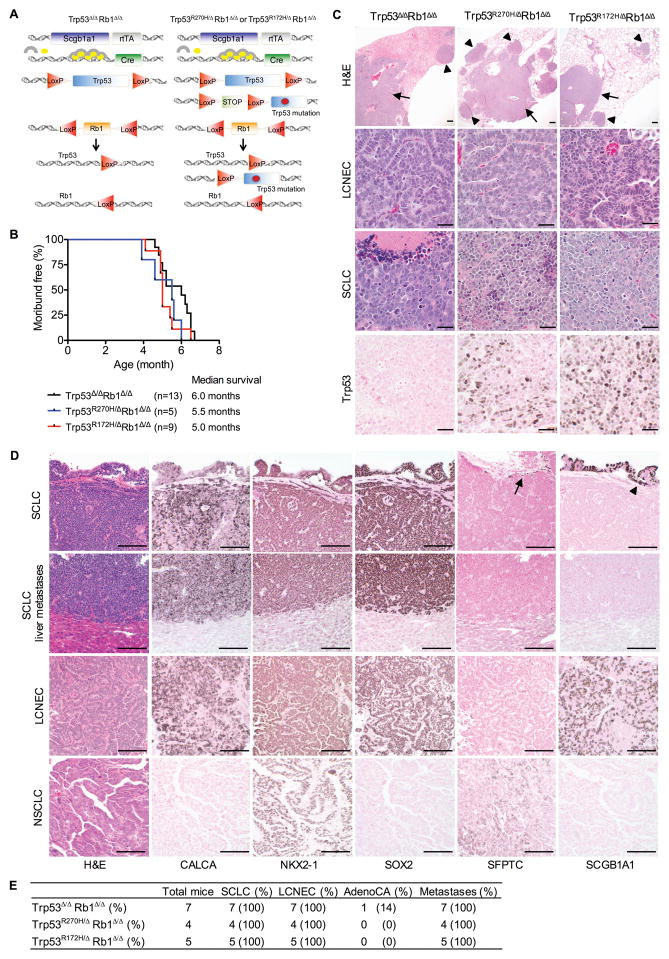
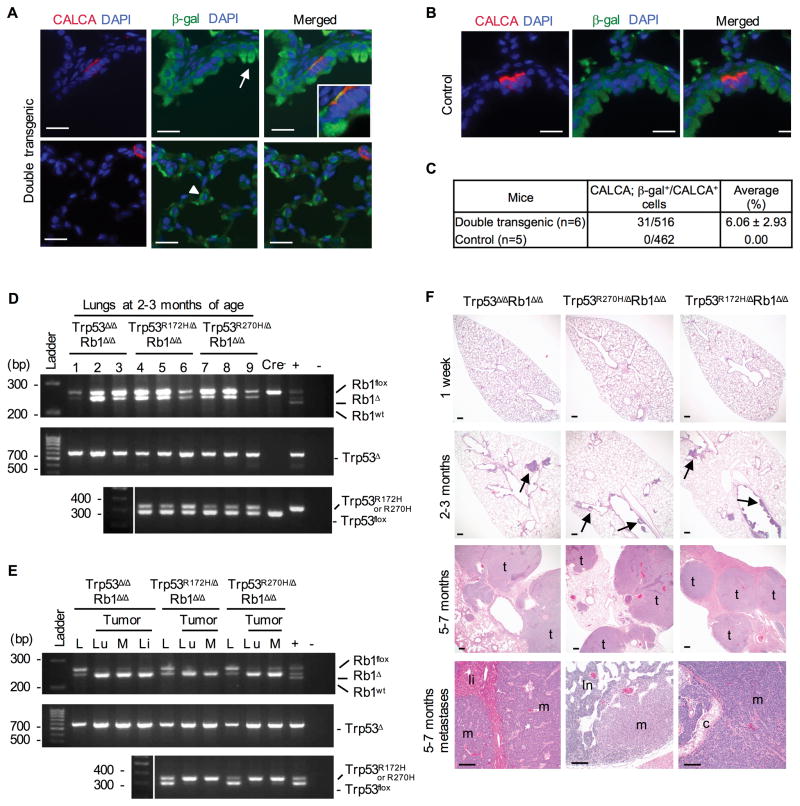
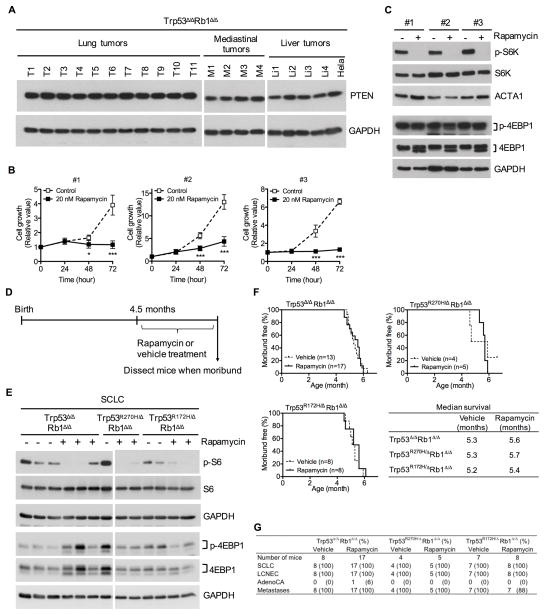
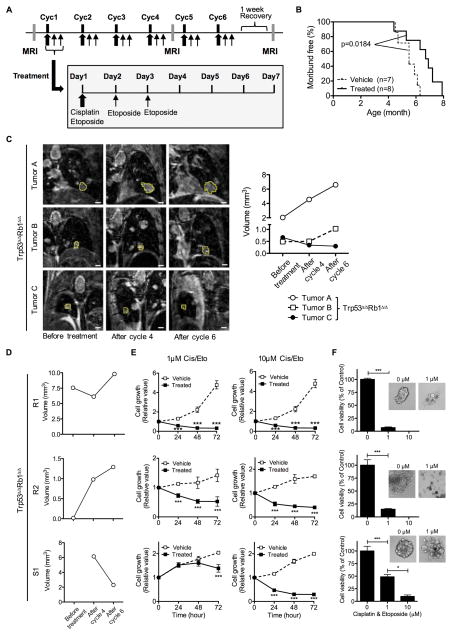
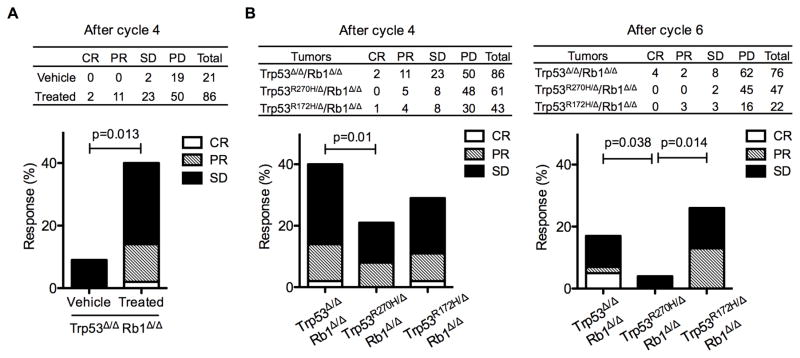
Lung cancer is the leading cause of cancer-related deaths with small-cell lung cancer (SCLC) as the most aggressive subtype. Preferential occurrence of TP53 missense mutations rather than loss implicates a selective advantage for TP53-mutant expression in SCLC pathogenesis. We show that lung epithelial expression of R270H and R172H (R273H and R175H in humans), common TRP53 mutants in lung cancer, combined with RB1 loss selectively results in two subtypes of neuroendocrine carcinoma, SCLC and large cell neuroendocrine carcinoma (LCNEC). Tumor initiation and progression occur in a remarkably consistent time frame with short latency and uniform progression to lethal metastatic disease by 7 months. R270H or R172H expression and TRP53 loss result in similar phenotypes demonstrating that TRP53 mutants promote lung carcinogenesis through loss-of-function and not gain-of-function mechanisms. Tumor responses to targeted and cytotoxic therapeutics were discordant in mice and corresponding tumor cell cultures demonstrating need to assess therapeutic response at the organismal level. Rapamycin did not have therapeutic efficacy in the mouse model despite inhibiting mTOR signaling and markedly suppressing tumor cell growth in culture. In contrast, cisplatin/etoposide treatment using a patient regimen prolonged survival with development of chemoresistance recapitulating human responses. R270H, but not R172H, expression conferred gain-of-function activity in attenuating chemotherapeutic efficacy. These data demonstrate a causative role for TRP53 mutants in development of chemoresistant lung cancer, and provide tractable preclinical models to test novel therapeutics for refractory disease. Mol Cancer Ther; 16(12); 2913-26. ©2017 AACR.
©2017 American Association for Cancer Research.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
