

Porphyromonas gingivalis Stimulates TLR2-PI3K Signaling to Escape Immune Clearance and Induce Bone Resorption Independently of MyD88
- PMID: 28848717
- PMCID: PMC5550410
- DOI: 10.3389/fcimb.2017.00359
Porphyromonas gingivalis Stimulates TLR2-PI3K Signaling to Escape Immune Clearance and Induce Bone Resorption Independently of MyD88
Abstract
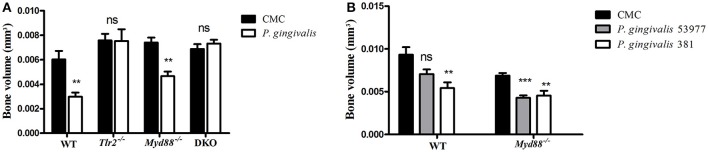
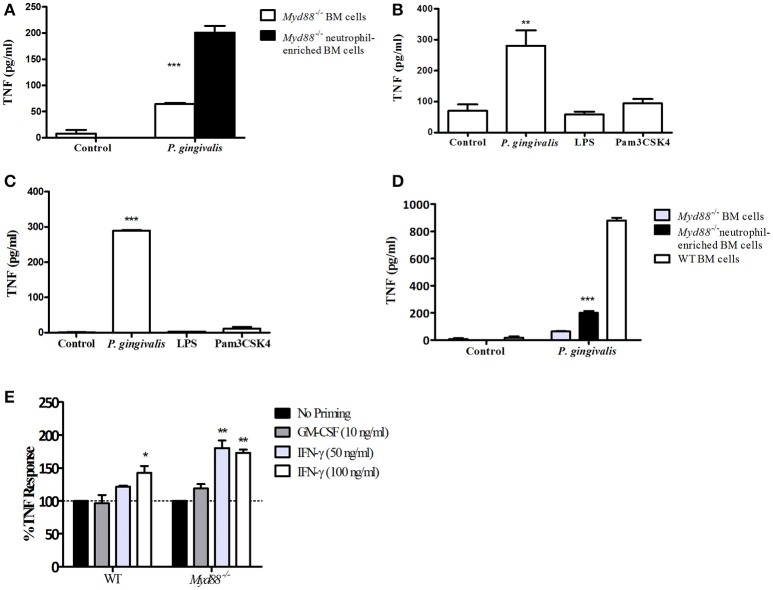
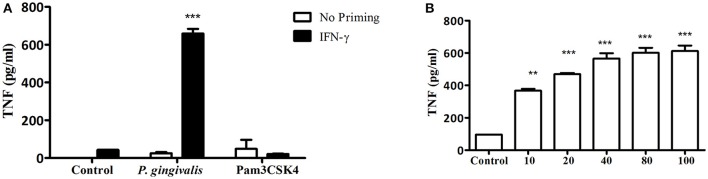
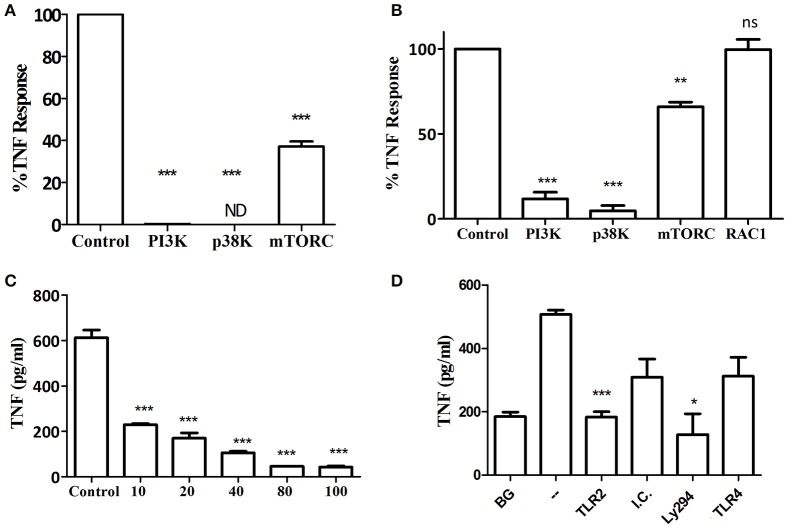
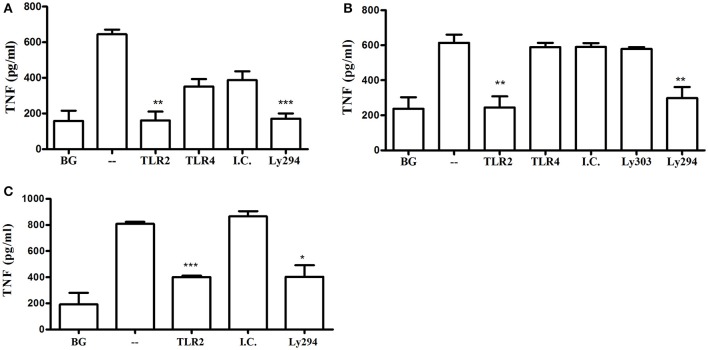
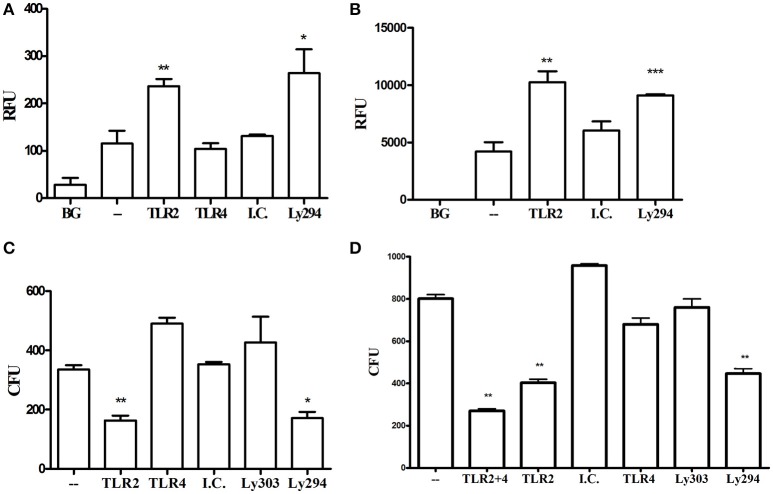
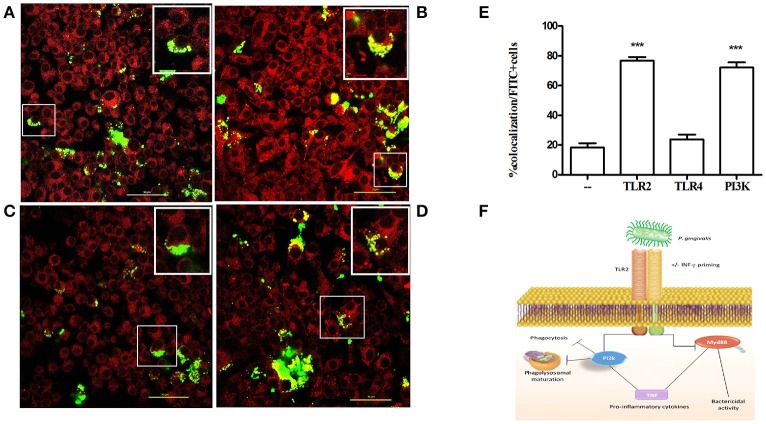
Porphyromonas gingivalis is a gram-negative anaerobic periodontal pathogen that persists in dysbiotic mixed-species biofilms alongside a dense inflammatory infiltrate of neutrophils and other leukocytes in the subgingival areas of the periodontium. Toll-like receptor 2 (TLR2) mediates the inflammatory response to P. gingivalis and TLR2-deficient mice resist alveolar bone resorption following oral challenge with this organism. Although, MyD88 is an adaptor protein considered necessary for TLR2-induced inflammation, we now report for the first time that oral challenge with P. gingivalis leads to alveolar bone resorption in the absence of MyD88. Indeed, in contrast to prototypical TLR2 agonists, such as the lipopeptide Pam3CSK4 that activates TLR2 in a strictly MyD88-dependent manner, P. gingivalis strikingly induced TLR2 signaling in neutrophils and macrophages regardless of the presence or absence of MyD88. Moreover, genetic or antibody-mediated inactivation of TLR2 completely reduced cytokine production in P. gingivalis-stimulated neutrophils or macrophages, suggesting that TLR2 plays a non-redundant role in the host response to P. gingivalis. In the absence of MyD88, inflammatory TLR2 signaling in P. gingivalis-stimulated neutrophils or macrophages depended upon PI3K. Intriguingly, TLR2-PI3K signaling was also critical to P. gingivalis evasion of killing by macrophages, since their ability to phagocytose this pathogen was reduced in a TLR2 and PI3K-dependent manner. Moreover, within those cells that did phagocytose bacteria, TLR2-PI3K signaling blocked phago-lysosomal maturation, thereby revealing a novel mechanism whereby P. gingivalis can enhance its intracellular survival. Therefore, P. gingivalis uncouples inflammation from bactericidal activity by substituting TLR2-PI3K in place of TLR2-MyD88 signaling. These findings further support the role of P. gingivalis as a keystone pathogen, which manipulates the host inflammatory response in a way that promotes bone loss but not bacterial clearance. Modulation of these host response factors may lead to novel therapeutic approaches to improve outcomes in disease conditions associated with P. gingivalis.
Keywords: MyD88; P. gingivalis; PI3 kinase; TLR2 signaling; immune evasion; macrophages; neutrophils.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases