

EL_PSSM-RT: DNA-binding residue prediction by integrating ensemble learning with PSSM Relation Transformation
- PMID: 28851273
- PMCID: PMC5576297
- DOI: 10.1186/s12859-017-1792-8
EL_PSSM-RT: DNA-binding residue prediction by integrating ensemble learning with PSSM Relation Transformation
Abstract
Background: Prediction of DNA-binding residue is important for understanding the protein-DNA recognition mechanism. Many computational methods have been proposed for the prediction, but most of them do not consider the relationships of evolutionary information between residues.
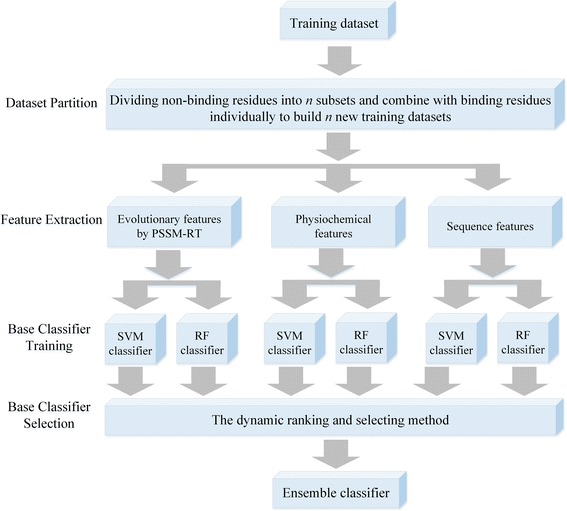
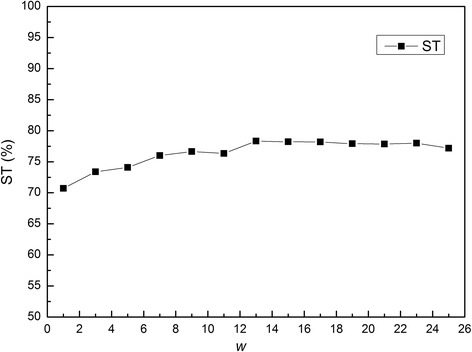
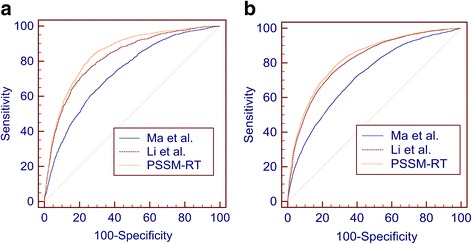
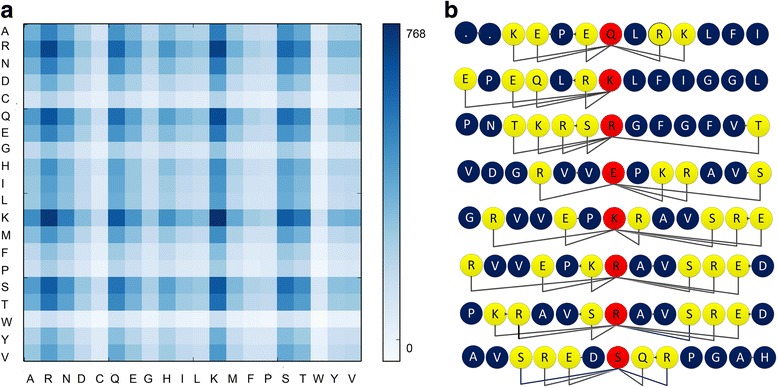
Results: In this paper, we first propose a novel residue encoding method, referred to as the Position Specific Score Matrix (PSSM) Relation Transformation (PSSM-RT), to encode residues by utilizing the relationships of evolutionary information between residues. PDNA-62 and PDNA-224 are used to evaluate PSSM-RT and two existing PSSM encoding methods by five-fold cross-validation. Performance evaluations indicate that PSSM-RT is more effective than previous methods. This validates the point that the relationship of evolutionary information between residues is indeed useful in DNA-binding residue prediction. An ensemble learning classifier (EL_PSSM-RT) is also proposed by combining ensemble learning model and PSSM-RT to better handle the imbalance between binding and non-binding residues in datasets. EL_PSSM-RT is evaluated by five-fold cross-validation using PDNA-62 and PDNA-224 as well as two independent datasets TS-72 and TS-61. Performance comparisons with existing predictors on the four datasets demonstrate that EL_PSSM-RT is the best-performing method among all the predicting methods with improvement between 0.02-0.07 for MCC, 4.18-21.47% for ST and 0.013-0.131 for AUC. Furthermore, we analyze the importance of the pair-relationships extracted by PSSM-RT and the results validates the usefulness of PSSM-RT for encoding DNA-binding residues.
Conclusions: We propose a novel prediction method for the prediction of DNA-binding residue with the inclusion of relationship of evolutionary information and ensemble learning. Performance evaluation shows that the relationship of evolutionary information between residues is indeed useful in DNA-binding residue prediction and ensemble learning can be used to address the data imbalance issue between binding and non-binding residues. A web service of EL_PSSM-RT ( http://hlt.hitsz.edu.cn:8080/PSSM-RT_SVM/ ) is provided for free access to the biological research community.
Keywords: DNA-binding residue; DNA-protein interaction; Ensemble learning; PSSM; Random forest; Relation transformation; SVM.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures


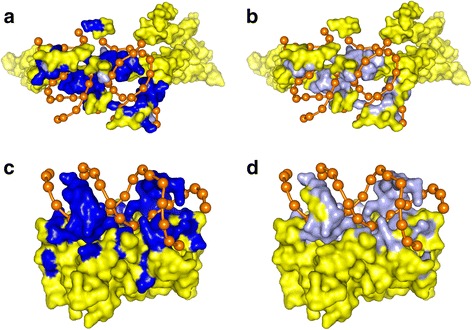
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
