

Gapless genome assembly of Colletotrichum higginsianum reveals chromosome structure and association of transposable elements with secondary metabolite gene clusters
- PMID: 28851275
- PMCID: PMC5576322
- DOI: 10.1186/s12864-017-4083-x
Gapless genome assembly of Colletotrichum higginsianum reveals chromosome structure and association of transposable elements with secondary metabolite gene clusters
Abstract
Background: The ascomycete fungus Colletotrichum higginsianum causes anthracnose disease of brassica crops and the model plant Arabidopsis thaliana. Previous versions of the genome sequence were highly fragmented, causing errors in the prediction of protein-coding genes and preventing the analysis of repetitive sequences and genome architecture.
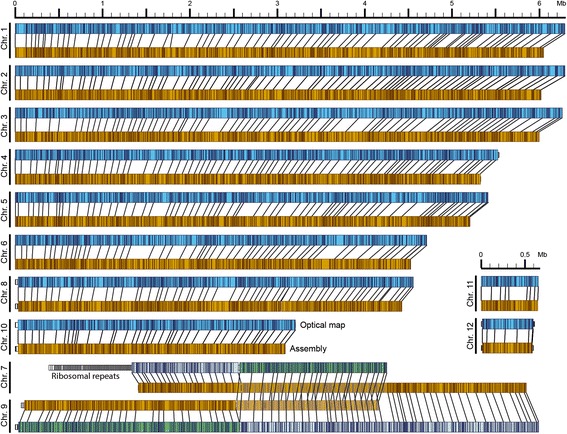
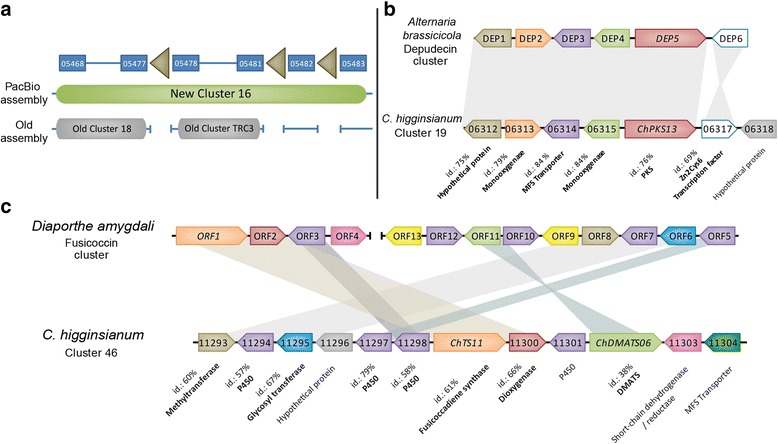
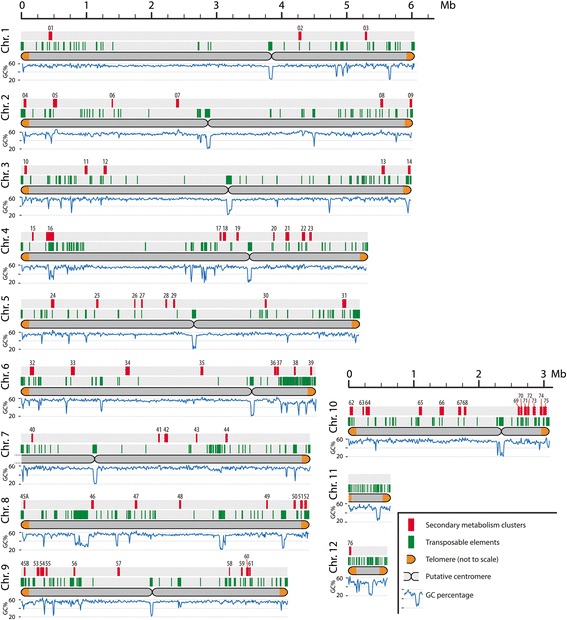
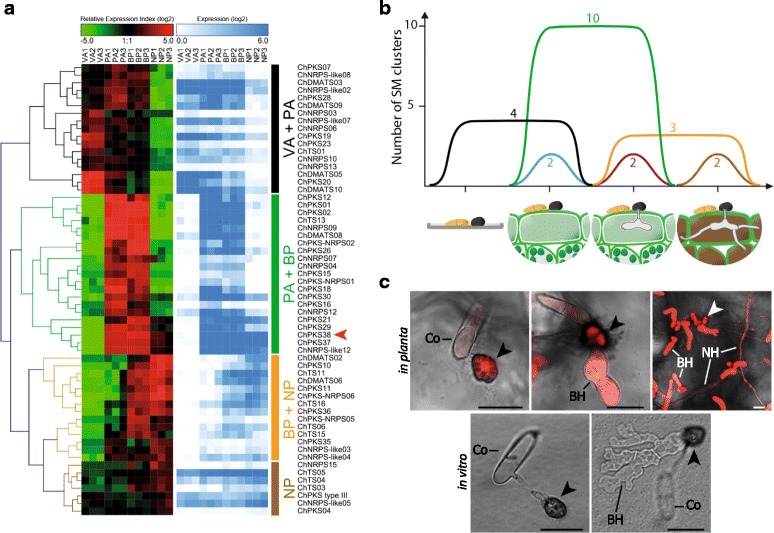
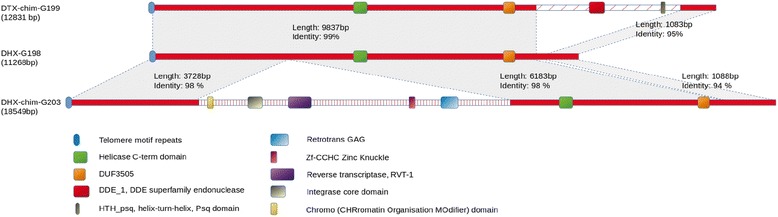
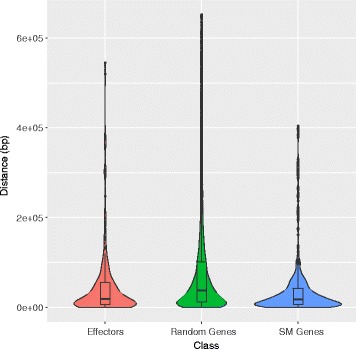
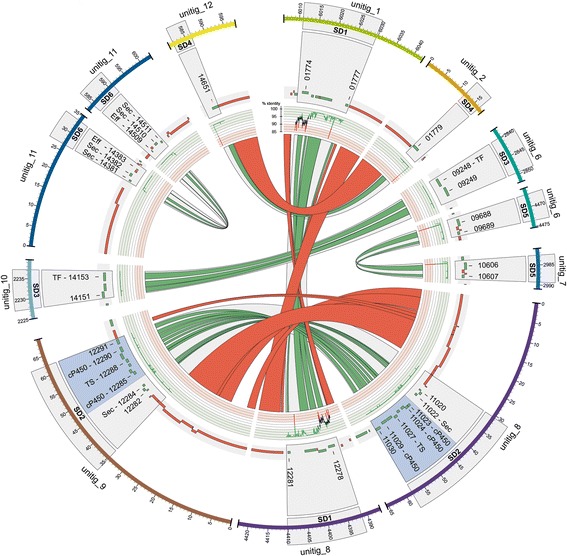
Results: Here, we re-sequenced the genome using single-molecule real-time (SMRT) sequencing technology and, in combination with optical map data, this provided a gapless assembly of all twelve chromosomes except for the ribosomal DNA repeat cluster on chromosome 7. The more accurate gene annotation made possible by this new assembly revealed a large repertoire of secondary metabolism (SM) key genes (89) and putative biosynthetic pathways (77 SM gene clusters). The two mini-chromosomes differed from the ten core chromosomes in being repeat- and AT-rich and gene-poor but were significantly enriched with genes encoding putative secreted effector proteins. Transposable elements (TEs) were found to occupy 7% of the genome by length. Certain TE families showed a statistically significant association with effector genes and SM cluster genes and were transcriptionally active at particular stages of fungal development. All 24 subtelomeres were found to contain one of three highly-conserved repeat elements which, by providing sites for homologous recombination, were probably instrumental in four segmental duplications.
Conclusion: The gapless genome of C. higginsianum provides access to repeat-rich regions that were previously poorly assembled, notably the mini-chromosomes and subtelomeres, and allowed prediction of the complete SM gene repertoire. It also provides insights into the potential role of TEs in gene and genome evolution and host adaptation in this asexual pathogen.
Keywords: Colletotrichum higginsianum; Fungal genome; SMRT sequencing; accessory chromosomes; optical map; secondary metabolism genes; segmental duplication; subtelomeres; transposable elements.
Conflict of interest statement
Authors information
The first two authors (J-FD and NL) contributed equally to this work.
Ethics approval and consent to participate
Seeds of the
Consent for publication
NA.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
The Landscape of Repetitive Elements in the Refined Genome of Chilli Anthracnose Fungus Colletotrichum truncatum.Front Microbiol. 2018 Oct 4;9:2367. doi: 10.3389/fmicb.2018.02367. eCollection 2018. Front Microbiol. 2018. PMID: 30337918 Free PMC article.
-
Complete genome of the Medicago anthracnose fungus, Colletotrichum destructivum, reveals a mini-chromosome-like region within a core chromosome.Microb Genom. 2024 Aug;10(8):001283. doi: 10.1099/mgen.0.001283. Microb Genom. 2024. PMID: 39166978 Free PMC article.
-
The landscape of transposable elements in the finished genome of the fungal wheat pathogen Mycosphaerella graminicola.BMC Genomics. 2014 Dec 17;15(1):1132. doi: 10.1186/1471-2164-15-1132. BMC Genomics. 2014. PMID: 25519841 Free PMC article.
-
Colletotrichum higginsianum as a Model for Understanding Host⁻Pathogen Interactions: A Review.Int J Mol Sci. 2018 Jul 23;19(7):2142. doi: 10.3390/ijms19072142. Int J Mol Sci. 2018. PMID: 30041456 Free PMC article. Review.
-
The Hidden Truths of Fungal Virulence and Adaptation on Hosts: Unraveling the Conditional Dispensability of Minichromosomes in the Hemibiotrophic Colletotrichum Pathogens.Int J Mol Sci. 2023 Dec 22;25(1):198. doi: 10.3390/ijms25010198. Int J Mol Sci. 2023. PMID: 38203369 Free PMC article. Review.
Cited by
-
Comparative Genomic Analyses of Colletotrichum lindemuthianum Pathotypes with Different Virulence Levels and Lifestyles.J Fungi (Basel). 2024 Sep 13;10(9):651. doi: 10.3390/jof10090651. J Fungi (Basel). 2024. PMID: 39330411 Free PMC article.
-
Colletotrichum shisoi sp. nov., an anthracnose pathogen of Perilla frutescens in Japan: molecular phylogenetic, morphological and genomic evidence.Sci Rep. 2019 Sep 16;9(1):13349. doi: 10.1038/s41598-019-50076-5. Sci Rep. 2019. PMID: 31527702 Free PMC article.
-
Chromosome rearrangements shape the diversification of secondary metabolism in the cyclosporin producing fungus Tolypocladium inflatum.BMC Genomics. 2019 Feb 7;20(1):120. doi: 10.1186/s12864-018-5399-x. BMC Genomics. 2019. PMID: 30732559 Free PMC article.
-
A Dispensable Chromosome Is Required for Virulence in the Hemibiotrophic Plant Pathogen Colletotrichum higginsianum.Front Microbiol. 2018 May 18;9:1005. doi: 10.3389/fmicb.2018.01005. eCollection 2018. Front Microbiol. 2018. PMID: 29867895 Free PMC article.
-
Plants under the Attack of Allies: Moving towards the Plant Pathobiome Paradigm.Plants (Basel). 2021 Jan 9;10(1):125. doi: 10.3390/plants10010125. Plants (Basel). 2021. PMID: 33435275 Free PMC article. Review.
References
-
- Seidl MF, Faino L, Shi-Kunne X, van den Berg GC, Bolton MD, Thomma BP. The Genome of the Saprophytic Fungus Verticillium tricorpus Reveals a Complex Effector Repertoire Resembling That of Its Pathogenic Relatives. Mol Plant Microbe Interact. 2015;28(3):362–373. doi: 10.1094/MPMI-06-14-0173-R. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
