

Using ALoFT to determine the impact of putative loss-of-function variants in protein-coding genes
- PMID: 28851873
- PMCID: PMC5575292
- DOI: 10.1038/s41467-017-00443-5
Using ALoFT to determine the impact of putative loss-of-function variants in protein-coding genes
Abstract
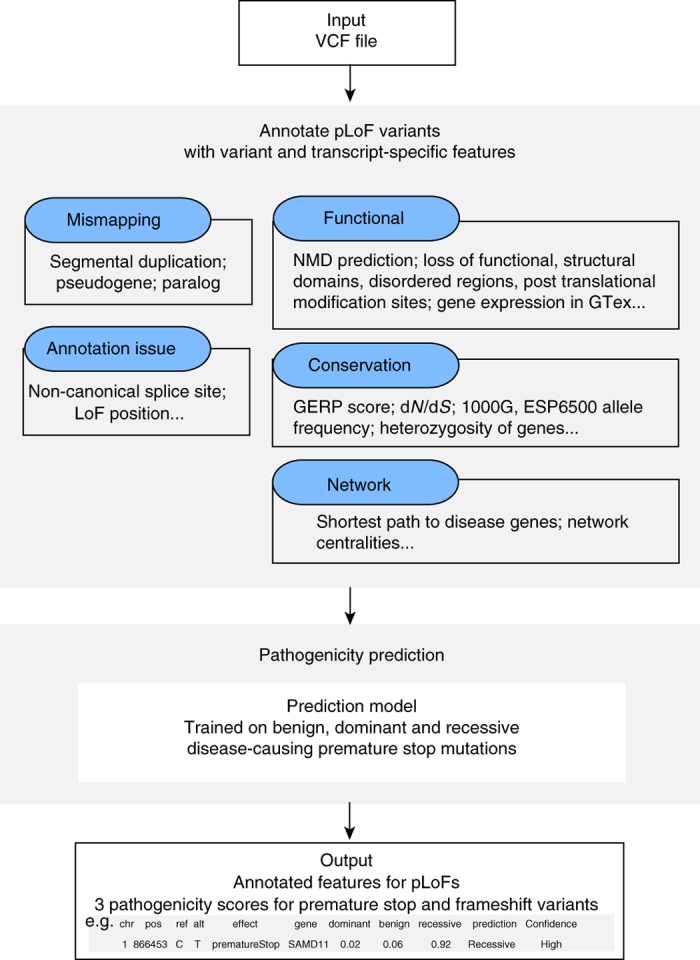
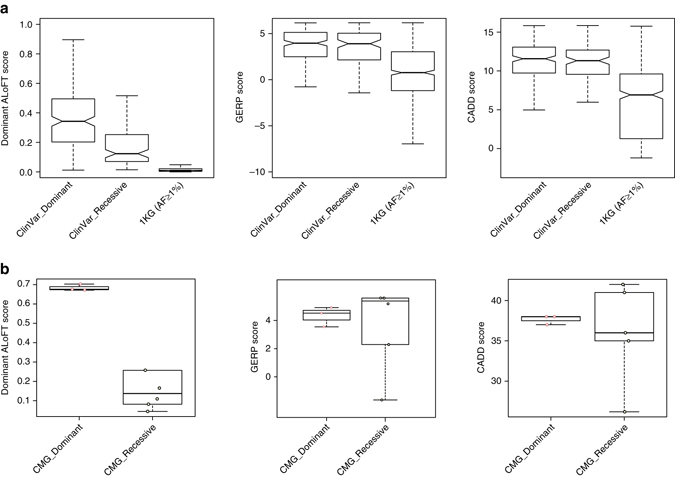
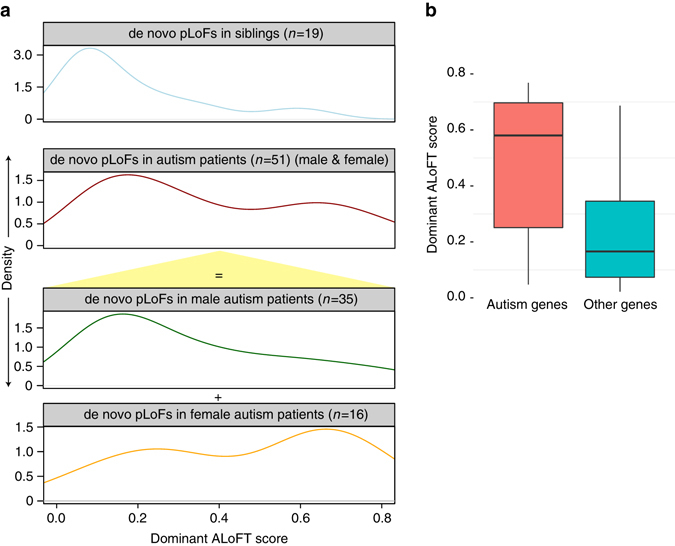
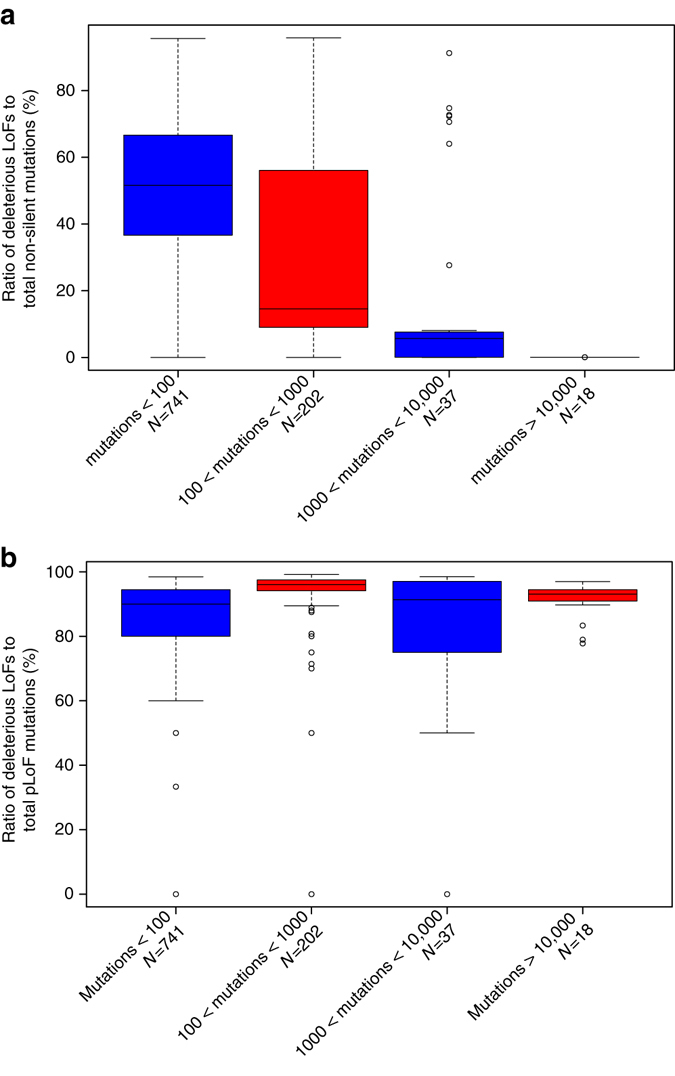
Variants predicted to result in the loss of function of human genes have attracted interest because of their clinical impact and surprising prevalence in healthy individuals. Here, we present ALoFT (annotation of loss-of-function transcripts), a method to annotate and predict the disease-causing potential of loss-of-function variants. Using data from Mendelian disease-gene discovery projects, we show that ALoFT can distinguish between loss-of-function variants that are deleterious as heterozygotes and those causing disease only in the homozygous state. Investigation of variants discovered in healthy populations suggests that each individual carries at least two heterozygous premature stop alleles that could potentially lead to disease if present as homozygotes. When applied to de novo putative loss-of-function variants in autism-affected families, ALoFT distinguishes between deleterious variants in patients and benign variants in unaffected siblings. Finally, analysis of somatic variants in >6500 cancer exomes shows that putative loss-of-function variants predicted to be deleterious by ALoFT are enriched in known driver genes.Variants causing loss of function (LoF) of human genes have clinical implications. Here, the authors present a method to predict disease-causing potential of LoF variants, ALoFT (annotation of Loss-of-Function Transcripts) and show its application to interpreting LoF variants in different contexts.
Conflict of interest statement
The authors declare no competing financial interests.
Figures


Similar articles
-
Deep sequencing of Danish Holstein dairy cattle for variant detection and insight into potential loss-of-function variants in protein coding genes.BMC Genomics. 2015 Dec 9;16:1043. doi: 10.1186/s12864-015-2249-y. BMC Genomics. 2015. PMID: 26645365 Free PMC article.
-
Postzygotic single-nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations.Hum Mutat. 2017 Aug;38(8):1002-1013. doi: 10.1002/humu.23255. Epub 2017 May 30. Hum Mutat. 2017. PMID: 28503910 Free PMC article.
-
Analysis of loss-of-function variants and 20 risk factor phenotypes in 8,554 individuals identifies loci influencing chronic disease.Nat Genet. 2015 Jun;47(6):640-2. doi: 10.1038/ng.3270. Epub 2015 Apr 27. Nat Genet. 2015. PMID: 25915599 Free PMC article.
-
Loss-of-function variants in the genomes of healthy humans.Hum Mol Genet. 2010 Oct 15;19(R2):R125-30. doi: 10.1093/hmg/ddq365. Epub 2010 Aug 30. Hum Mol Genet. 2010. PMID: 20805107 Free PMC article. Review.
-
The genetics of autism.Pediatrics. 2004 May;113(5):e472-86. doi: 10.1542/peds.113.5.e472. Pediatrics. 2004. PMID: 15121991 Review.
Cited by
-
VIPdb, a genetic Variant Impact Predictor Database.Hum Mutat. 2019 Sep;40(9):1202-1214. doi: 10.1002/humu.23858. Epub 2019 Aug 17. Hum Mutat. 2019. PMID: 31283070 Free PMC article.
-
Human subsistence and signatures of selection on chemosensory genes.Commun Biol. 2023 Jul 3;6(1):683. doi: 10.1038/s42003-023-05047-y. Commun Biol. 2023. PMID: 37400713 Free PMC article.
-
The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy.Nat Genet. 2019 Nov;51(11):1645-1651. doi: 10.1038/s41588-019-0517-5. Epub 2019 Oct 28. Nat Genet. 2019. PMID: 31659324 Free PMC article.
-
FAVOR: functional annotation of variants online resource and annotator for variation across the human genome.Nucleic Acids Res. 2023 Jan 6;51(D1):D1300-D1311. doi: 10.1093/nar/gkac966. Nucleic Acids Res. 2023. PMID: 36350676 Free PMC article.
-
WWP1 Gain-of-Function Inactivation of PTEN in Cancer Predisposition.N Engl J Med. 2020 May 28;382(22):2103-2116. doi: 10.1056/NEJMoa1914919. N Engl J Med. 2020. PMID: 32459922 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
