

Ape1 guides DNA repair pathway choice that is associated with drug tolerance in glioblastoma
- PMID: 28852018
- PMCID: PMC5574897
- DOI: 10.1038/s41598-017-10013-w
Ape1 guides DNA repair pathway choice that is associated with drug tolerance in glioblastoma
Abstract
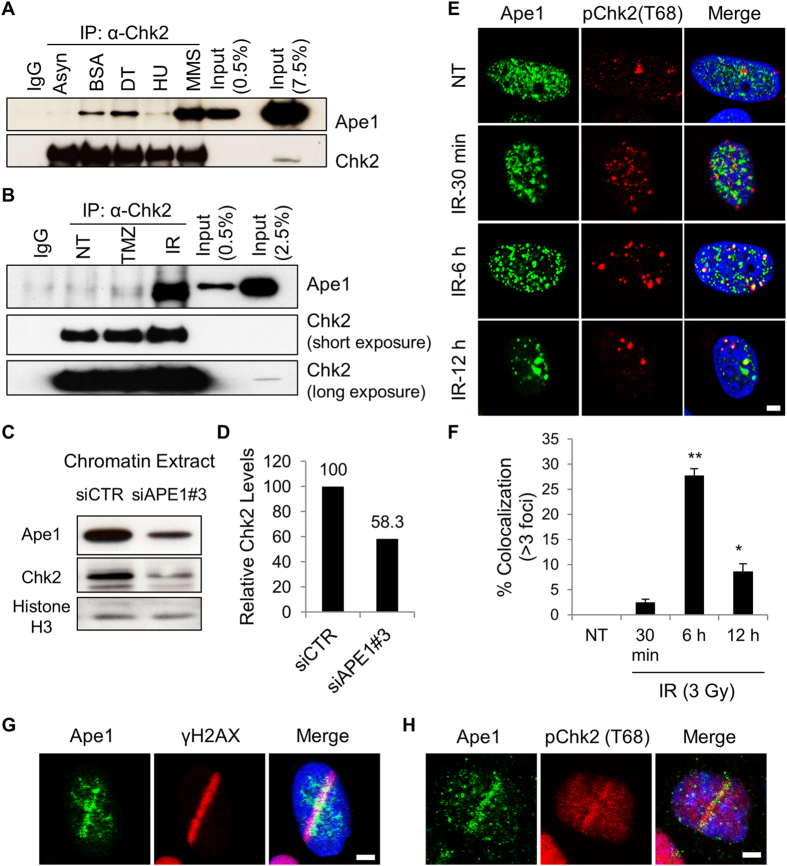
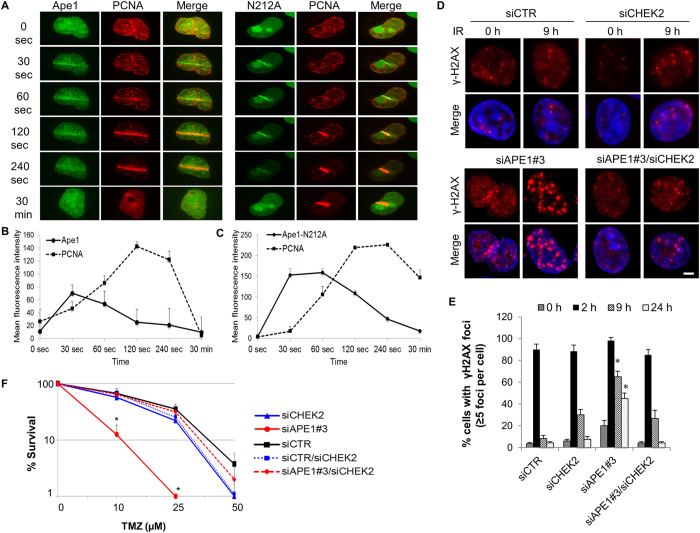
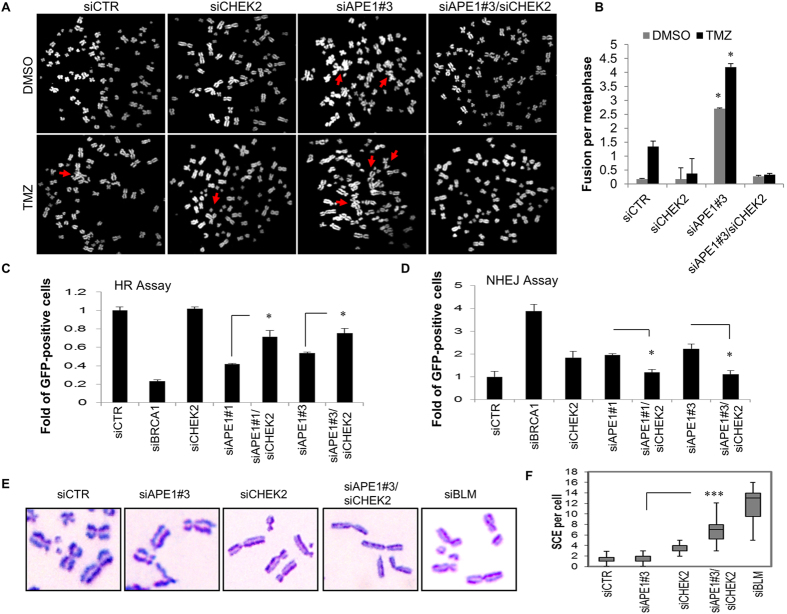
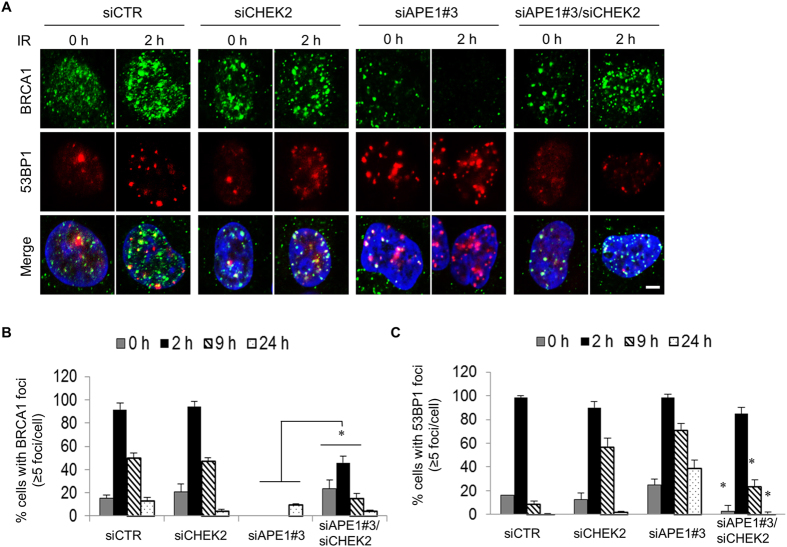
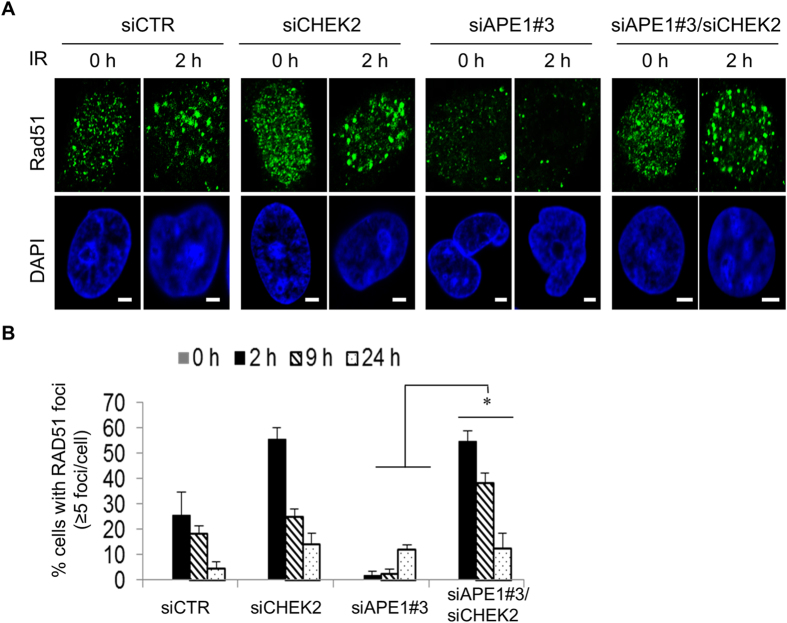
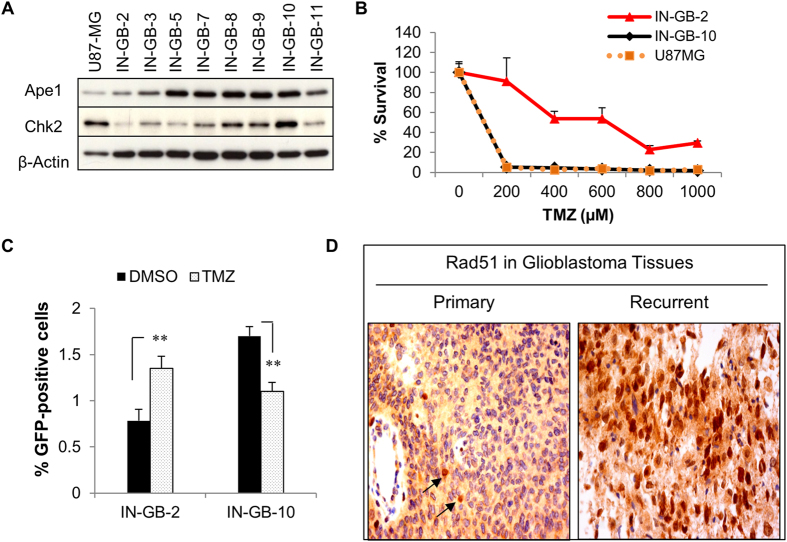
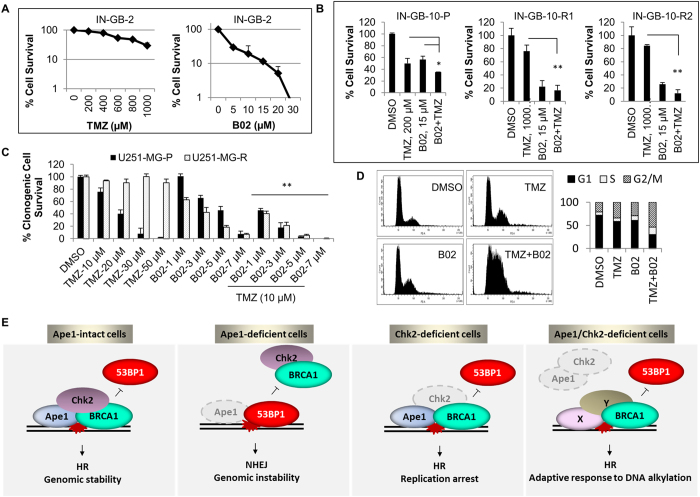
Ape1 is the major apurinic/apyrimidinic (AP) endonuclease activity in mammalian cells, and a key factor in base-excision repair of DNA. High expression or aberrant subcellular distribution of Ape1 has been detected in many cancer types, correlated with drug response, tumor prognosis, or patient survival. Here we present evidence that Ape1 facilitates BRCA1-mediated homologous recombination repair (HR), while counteracting error-prone non-homologous end joining of DNA double-strand breaks. Furthermore, Ape1, coordinated with checkpoint kinase Chk2, regulates drug response of glioblastoma cells. Suppression of Ape1/Chk2 signaling in glioblastoma cells facilitates alternative means of damage site recruitment of HR proteins as part of a genomic defense system. Through targeting "HR-addicted" temozolomide-resistant glioblastoma cells via a chemical inhibitor of Rad51, we demonstrated that targeting HR is a promising strategy for glioblastoma therapy. Our study uncovers a critical role for Ape1 in DNA repair pathway choice, and provides a mechanistic understanding of DNA repair-supported chemoresistance in glioblastoma cells.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells.J Pharmacol Exp Ther. 2010 Sep 1;334(3):988-98. doi: 10.1124/jpet.110.169128. Epub 2010 May 26. J Pharmacol Exp Ther. 2010. PMID: 20504914 Free PMC article.
-
Oxidative DNA damage is concurrently repaired by base excision repair (BER) and apyrimidinic endonuclease 1 (APE1)-initiated nonhomologous end joining (NHEJ) in cortical neurons.Neuropathol Appl Neurobiol. 2020 Jun;46(4):375-390. doi: 10.1111/nan.12584. Epub 2019 Nov 6. Neuropathol Appl Neurobiol. 2020. PMID: 31628877 Free PMC article.
-
The role of the N-terminal domain of human apurinic/apyrimidinic endonuclease 1, APE1, in DNA glycosylase stimulation.DNA Repair (Amst). 2018 Apr;64:10-25. doi: 10.1016/j.dnarep.2018.02.001. Epub 2018 Feb 11. DNA Repair (Amst). 2018. PMID: 29475157
-
AP Endonuclease 1 as a Key Enzyme in Repair of Apurinic/Apyrimidinic Sites.Biochemistry (Mosc). 2016 Sep;81(9):951-67. doi: 10.1134/S0006297916090042. Biochemistry (Mosc). 2016. PMID: 27682167 Review.
-
The multifunctional APE1 DNA repair-redox signaling protein as a drug target in human disease.Drug Discov Today. 2021 Jan;26(1):218-228. doi: 10.1016/j.drudis.2020.10.015. Epub 2020 Oct 24. Drug Discov Today. 2021. PMID: 33148489 Free PMC article. Review.
Cited by
-
Role of apurinic/apyrimidinic nucleases in the regulation of homologous recombination in myeloma: mechanisms and translational significance.Blood Cancer J. 2018 Sep 25;8(10):92. doi: 10.1038/s41408-018-0129-9. Blood Cancer J. 2018. PMID: 30301882 Free PMC article.
-
APE1 distinguishes DNA substrates in exonucleolytic cleavage by induced space-filling.Nat Commun. 2021 Jan 27;12(1):601. doi: 10.1038/s41467-020-20853-2. Nat Commun. 2021. PMID: 33504804 Free PMC article.
-
Shining light on the response to repair intermediates in DNA of living cells.DNA Repair (Amst). 2020 Jan;85:102749. doi: 10.1016/j.dnarep.2019.102749. Epub 2019 Nov 12. DNA Repair (Amst). 2020. PMID: 31790865 Free PMC article.
-
Targets for Renal Carcinoma Growth Control Identified by Screening FOXD1 Cell Proliferation Pathways.Cancers (Basel). 2022 Aug 16;14(16):3958. doi: 10.3390/cancers14163958. Cancers (Basel). 2022. PMID: 36010951 Free PMC article.
-
The DNA Double-Strand Break Repair in Glioma: Molecular Players and Therapeutic Strategies.Mol Neurobiol. 2022 Sep;59(9):5326-5365. doi: 10.1007/s12035-022-02915-2. Epub 2022 Jun 13. Mol Neurobiol. 2022. PMID: 35696013 Review.
References
-
- Nakamura J, et al. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res. 1998;58:222–225. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
