

Beyond editing to writing large genomes
- PMID: 28852223
- PMCID: PMC5793211
- DOI: 10.1038/nrg.2017.59
Beyond editing to writing large genomes
Abstract
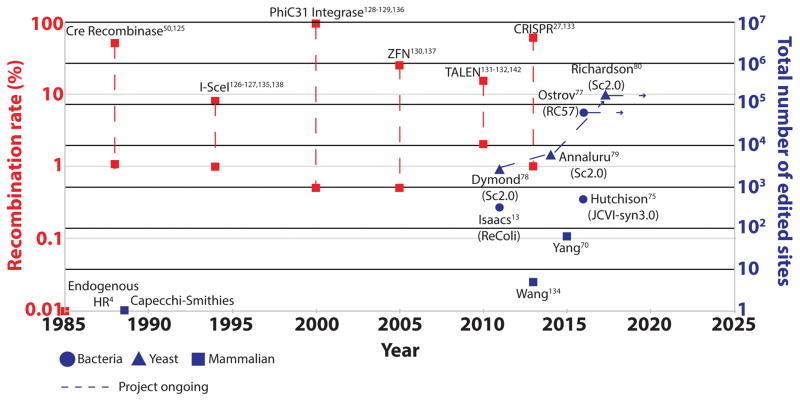
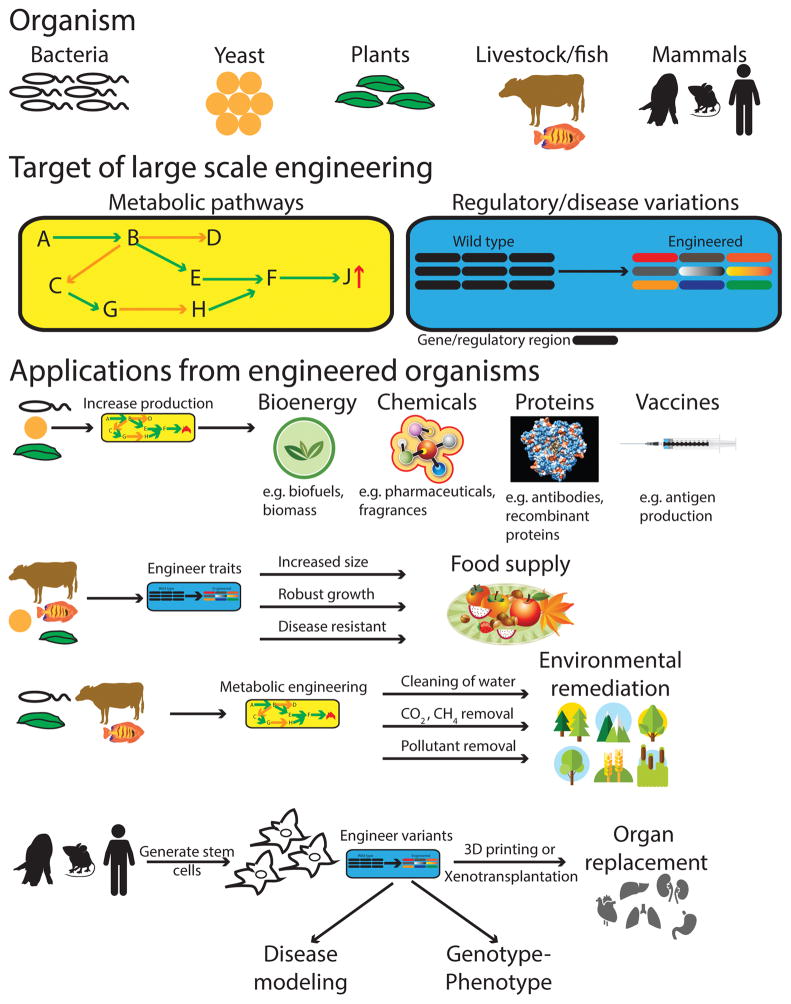
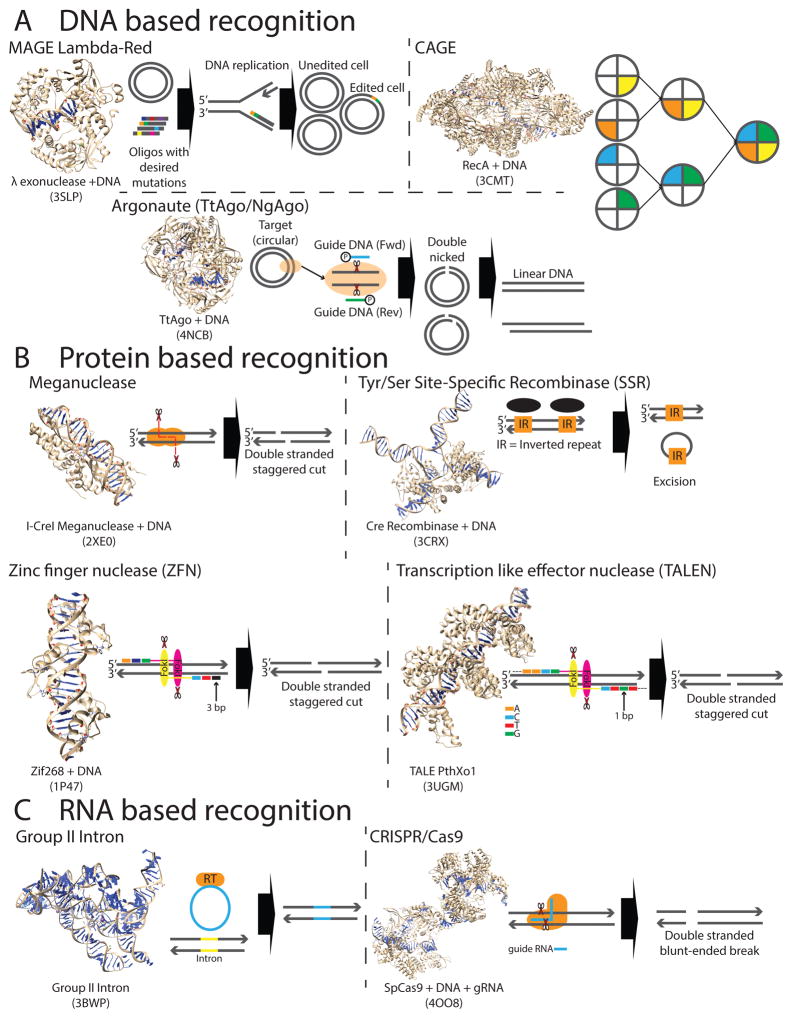
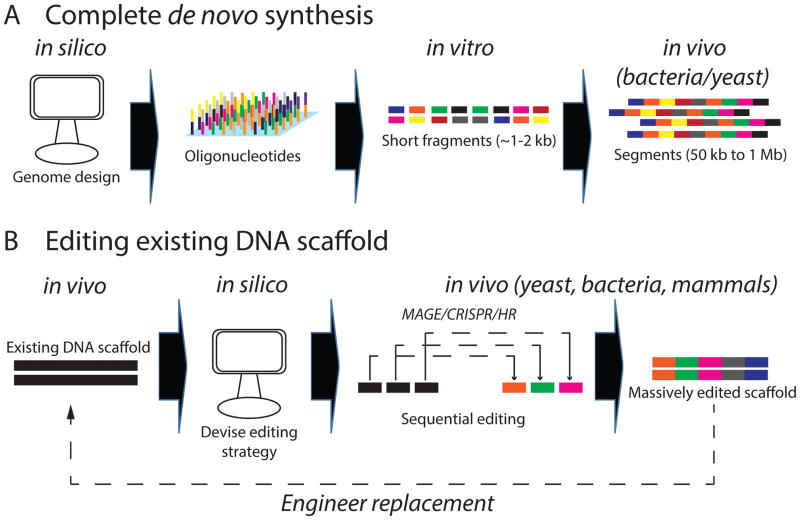
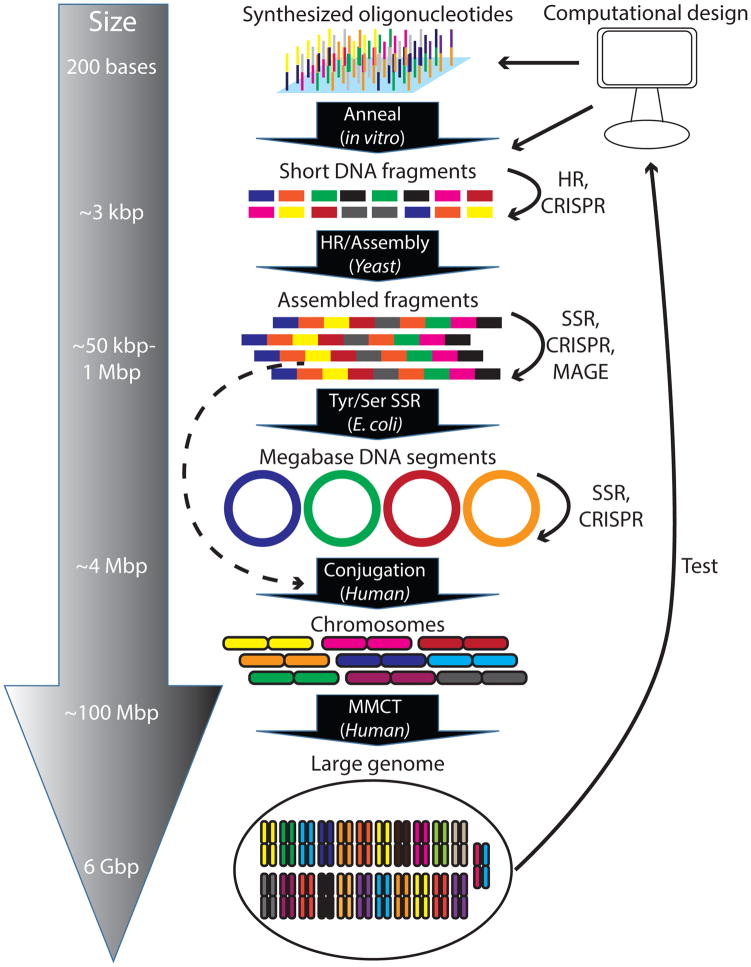
Recent exponential advances in genome sequencing and engineering technologies have enabled an unprecedented level of interrogation into the impact of DNA variation (genotype) on cellular function (phenotype). Furthermore, these advances have also prompted realistic discussion of writing and radically re-writing complex genomes. In this Perspective, we detail the motivation for large-scale engineering, discuss the progress made from such projects in bacteria and yeast and describe how various genome-engineering technologies will contribute to this effort. Finally, we describe the features of an ideal platform and provide a roadmap to facilitate the efficient writing of large genomes.
Conflict of interest statement
G.M.C. is a co-founder of Editas Medicine and eGenesis Bio and serves advisory roles in several companies involved in genome editing and engineering. A detailed listing of G.M.C.’s Tech Transfer, Advisory Roles, and Funding Sources can be obtained from
Figures
References
-
- Smithies O, Gregg RG, Boggs SS, Koralewski MA, Kucherlapati RS. Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature. 1985;317:230–234. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
