

Ataxia-pancytopenia syndrome with SAMD9L mutations
- PMID: 28852709
- PMCID: PMC5570676
- DOI: 10.1212/NXG.0000000000000183
Ataxia-pancytopenia syndrome with SAMD9L mutations
Abstract
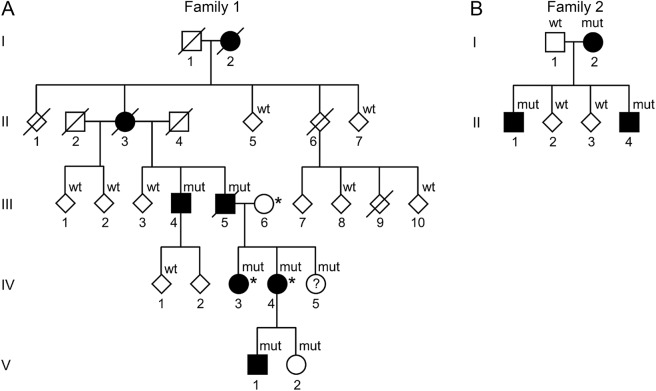
Objective: We describe the neurologic, neuroradiologic, and ophthalmologic phenotype of 1 Swedish and 1 Finnish family with autosomal dominant ataxia-pancytopenia (ATXPC) syndrome and SAMD9L mutations.
Methods: Members of these families with germline SAMD9L c.2956C>T, p.Arg986Cys, or c.2672T>C, p.Ile891Thr mutations underwent structured interviews and neurologic and ophthalmologic examinations. Neuroimaging was performed, and medical records were reviewed. Previous publications on SAMD9L-ATXPC were reviewed.
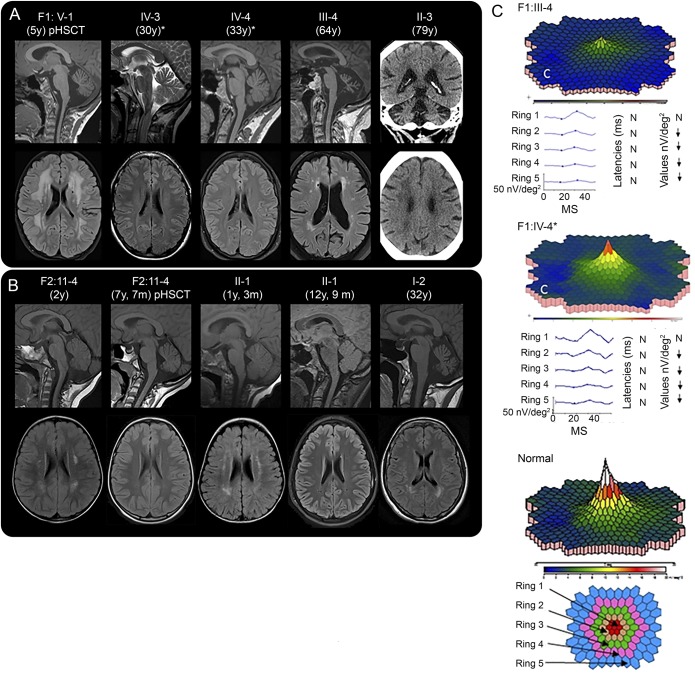
Results: Twelve individuals in both families were affected clinically. All mutation carriers examined had balance impairment, although severity was very variable. All but 1 had nystagmus, and all but 1 had pyramidal tract signs. Neurologic features were generally present from childhood on and progressed slowly. Two adult patients, who experienced increasing clumsiness, glare, and difficulties with gaze fixation, had paracentral retinal dysfunction verified by multifocal electroretinography. Brain MRI showed early, marked cerebellar atrophy in most carriers and variable cerebral periventricular white matter T2 hyperintensities. Two children were treated with hematopoietic stem cell transplantation for hematologic malignancies, and the neurologic symptoms of one of these worsened after treatment. Three affected individuals had attention deficit hyperactivity disorder or cognitive problems. Retinal dysfunction was not previously reported in individuals with ATXPC.
Conclusions: The neurologic phenotype of this syndrome is defined by balance or gait impairment, nystagmus, hyperreflexia in the lower limbs and, frequently, marked cerebellar atrophy. Paracentral retinal dysfunction may contribute to glare, reading problems, and clumsiness. Timely diagnosis of ATXPC is important to address the risk for severe hemorrhage, infection, and hematologic malignancies inherent in this syndrome; regular hematologic follow-up might be beneficial.
Figures
References
-
- Li FP, Potter NU, Buchanan GR, Vawter G, Whang-Peng J, Rosen RB. A family with acute leukemia, hypoplastic anemia and cerebellar ataxia: association with bone marrow C-monosomy. Am J Med 1978;65:933–940. - PubMed
-
- Li FP, Hecht F, Kaiser-McCaw B, Baranko PV, Potter NU. Ataxia-pancytopenia: syndrome of cerebellar ataxia, hypoplastic anemia, monosomy 7, and acute myelogenous leukemia. Cancer Genet Cytogenet 1981;4:189–196. - PubMed
-
- Daghistani D, Curless R, Toledano SR, Ayyar DR. Ataxia-pancytopenia and monosomy 7 syndrome. J Pediatr 1989;115:108–110. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources