

Cystic Fibrosis
- PMID: 28855057
- PMCID: PMC5596161
- DOI: 10.3238/arztebl.2017.0564
Cystic Fibrosis
Abstract
Background: Universal screening of newborn babies for cystic fibrosis was launched in Germany on 1 September 2016. Here we present up-to-date information on the diagnosis, treatment, and prognosis of this disease.
Methods: This article is based on relevant publications retrieved by a selective search in PubMed, along with guidelines from Germany and abroad and systematic reviews.
Results: Cystic fibrosis is caused by a gene mutation leading to dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) protein. It affects multiple organ systems-the lungs, pancreas, upper airways, liver, intestine, and reproductive organs-to varying degrees. Its incidence among newborn babies in Germany is between 1 in 3300 and 1 in 4800. Its diagnosis requires both clinical evidence (positive newborn screening, sibling[s] with cystic fibrosis, clinical signs) and the demonstration of CFTR dysfunction by an elevated chloride concentration in sweat, and/or two disease-causing mutations, and/or abnormal electrophysiological findings (nasal potential difference measurement, intestinal short-circuit current measurement). Patients should be cared for by specialized cystic fibrosis centers in close cooperation with their primary care physicians. The median life span of patients with this disease has risen steadily to the current value of 40 years. Aside from symptomatic treatment, the first mutation- specific treatments have recently become available.
Conclusion: Early diagnosis and optimized treatment prolong the lives of persons with cystic fibrosis and improve their quality of life. Causally directed treatment for all patients and their effects on the course of disease are now central issues for further research.
Figures
References
-
- World Health Organization. The molecular genetic epidemiology of cystic fibrosis. www.who.int/genomics/publications/reports/en/index.html (last accessed on 14 February 2017)
-
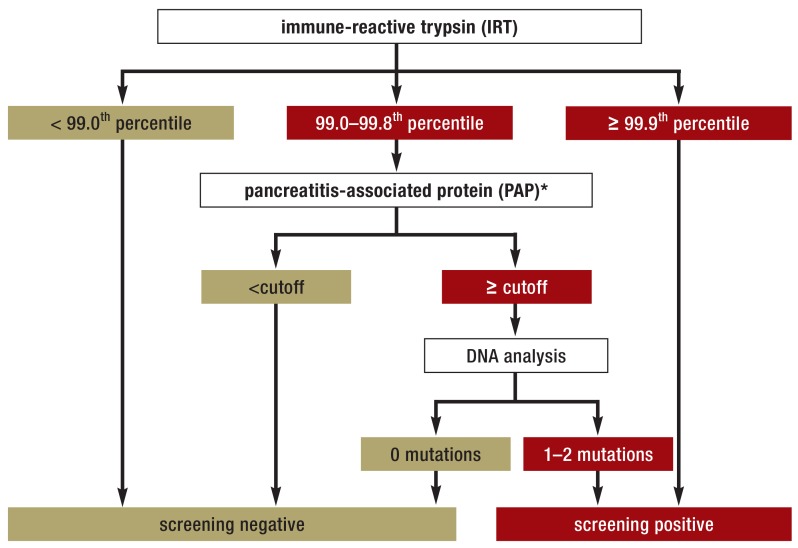
- Sommerburg O, Hammermann J, Lindner M, et al. Five years of experience with biochemical cystic fibrosis newborn screening based on IRT/PAP in Germany. Pediatr Pulmonol. 2015;50:655–664. - PubMed
-
- Sens B, Stern M. Qualitätssicherung Mukoviszidose 2012. Bad Honnef: Hippocampus Verlag. 2013
-
- Nährlich L, Burkhart M, Wiese B. Deutsches Mukoviszidoseregister- Berichtsband 2015. www.muko.info/fileadmin/redaktion/datei_gruppen/muko_institut/Register/B... (last accessed on 10 December 2016
-
- Gemeinsamer Bundesausschuss. Kinder-Richtlinie: Formale und inhaltliche Überarbeitung (Neustrukturierung) - Neufassung. www.g-ba.de/informationen/beschluesse/2287/ (last accessed on 10 September 2016)
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical