

The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology
- PMID: 28855259
- PMCID: PMC5668948
- DOI: 10.1101/gr.218032.116
The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology
Abstract
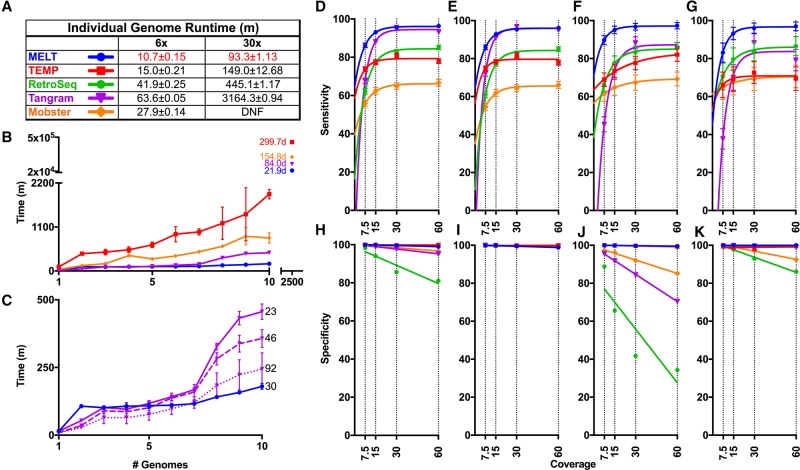
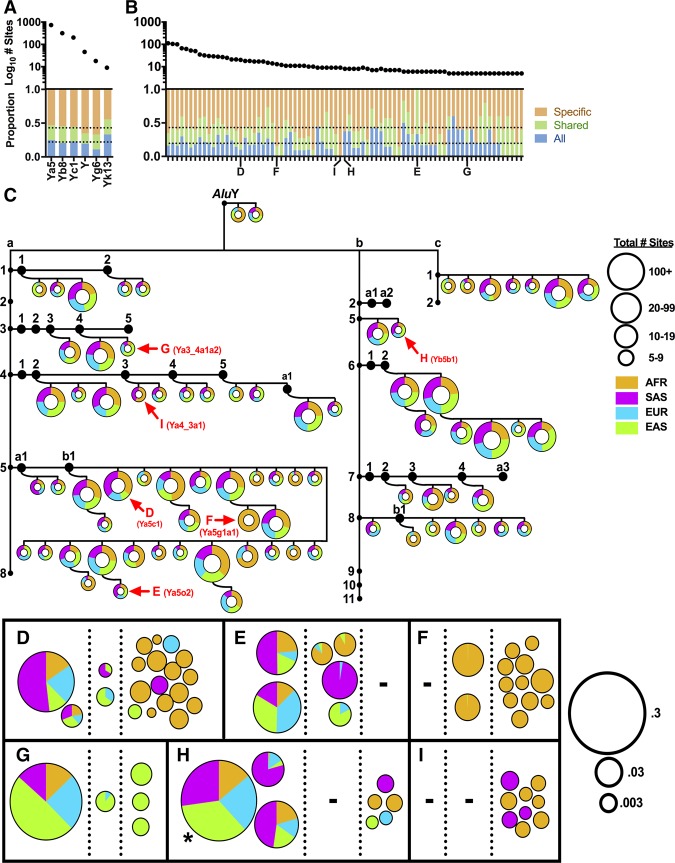
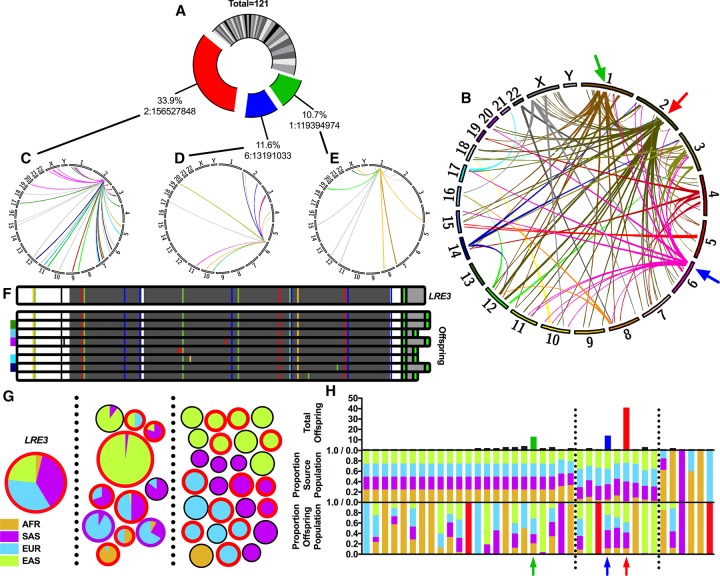
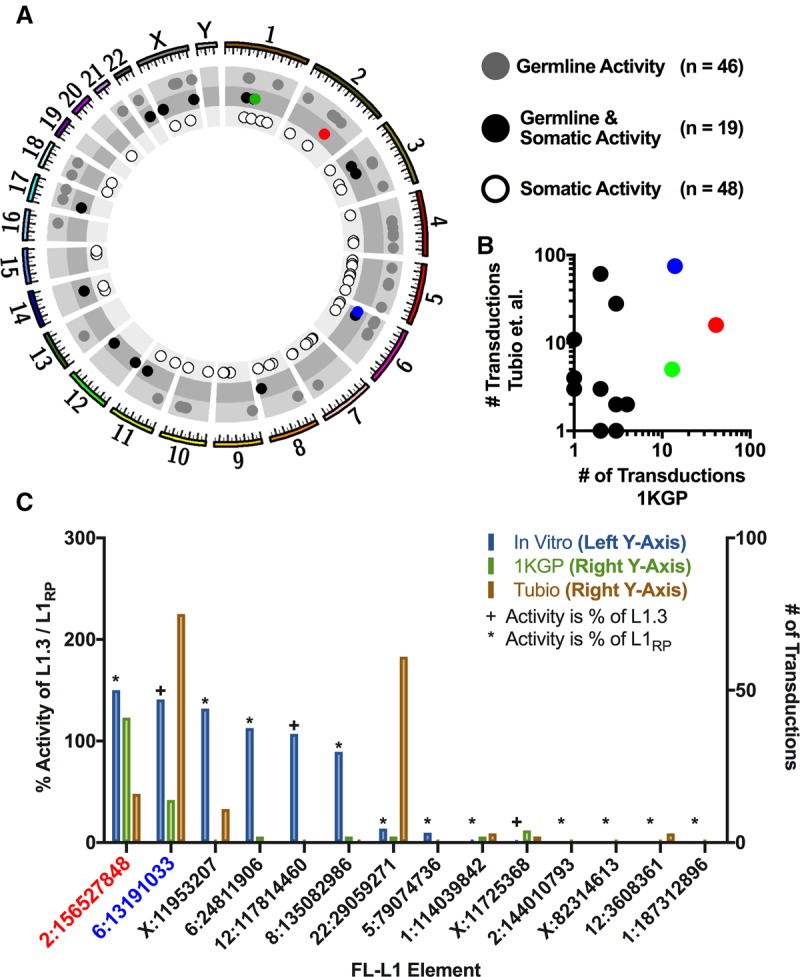
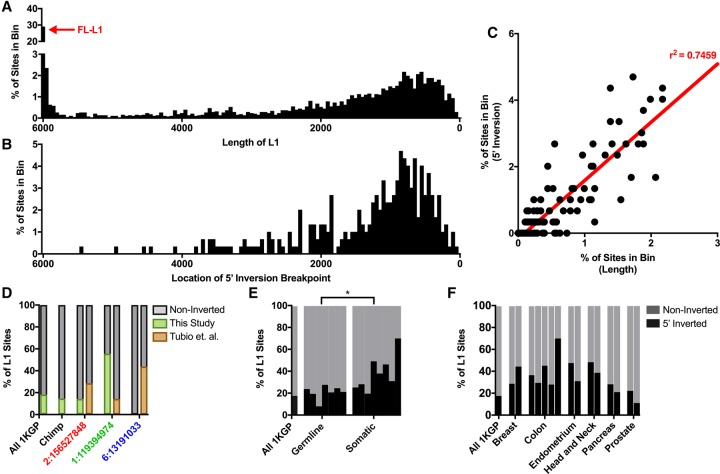
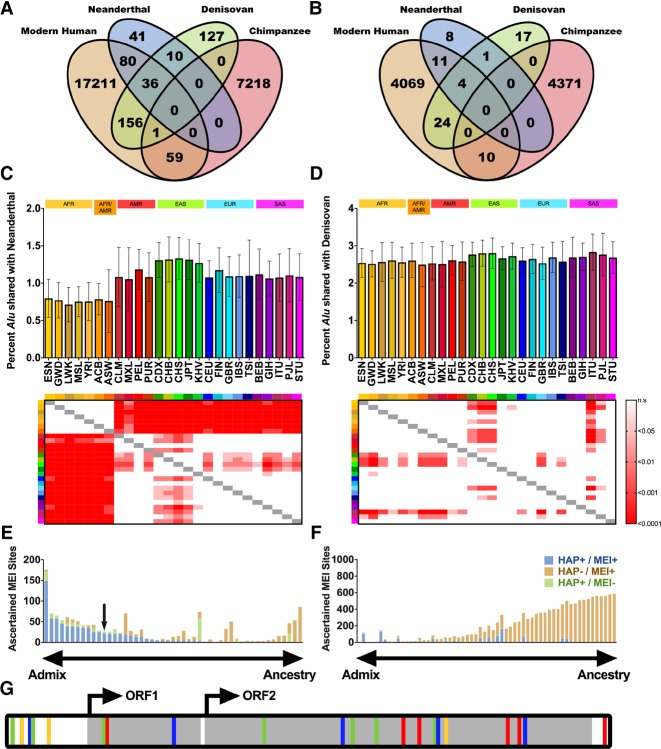
Mobile element insertions (MEIs) represent ∼25% of all structural variants in human genomes. Moreover, when they disrupt genes, MEIs can influence human traits and diseases. Therefore, MEIs should be fully discovered along with other forms of genetic variation in whole genome sequencing (WGS) projects involving population genetics, human diseases, and clinical genomics. Here, we describe the Mobile Element Locator Tool (MELT), which was developed as part of the 1000 Genomes Project to perform MEI discovery on a population scale. Using both Illumina WGS data and simulations, we demonstrate that MELT outperforms existing MEI discovery tools in terms of speed, scalability, specificity, and sensitivity, while also detecting a broader spectrum of MEI-associated features. Several run modes were developed to perform MEI discovery on local and cloud systems. In addition to using MELT to discover MEIs in modern humans as part of the 1000 Genomes Project, we also used it to discover MEIs in chimpanzees and ancient (Neanderthal and Denisovan) hominids. We detected diverse patterns of MEI stratification across these populations that likely were caused by (1) diverse rates of MEI production from source elements, (2) diverse patterns of MEI inheritance, and (3) the introgression of ancient MEIs into modern human genomes. Overall, our study provides the most comprehensive map of MEIs to date spanning chimpanzees, ancient hominids, and modern humans and reveals new aspects of MEI biology in these lineages. We also demonstrate that MELT is a robust platform for MEI discovery and analysis in a variety of experimental settings.
© 2017 Gardner et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Batzer MA, Deininger PL. 2002. Alu repeats and human genomic diversity. Nat Rev Genet 3: 370–379. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases